Three-dimensional chromatin architecture datasets for aging and alzheimer’s disease
Three-dimensional chromatin architecture datasets for aging and alzheimer’s disease"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Recently, increasing studies are indicating a close association between dysregulated enhancers and neurodegenerative diseases, such as Alzheimer’s disease (AD). However, their
contributions were poorly defined for lacking direct links to disease genes. To bridge this gap, we presented the Hi-C datasets of 4 AD patients, 4 dementia-free aged and 3 young subjects,
including 30 billion reads. As applications, we utilized them to link the AD risk SNPs and dysregulated epigenetic marks to the target genes. Combining with epigenetic data, we observed more
detailed interactions among regulatory regions and found that many known AD risk genes were under long-distance promoter-enhancer interactions. For future AD and aging studies, our datasets
provide a reference landscape to better interpret findings of association and epigenetic studies for AD and aging process. SIMILAR CONTENT BEING VIEWED BY OTHERS THE THREE-DIMENSIONAL
LANDSCAPE OF CORTICAL CHROMATIN ACCESSIBILITY IN ALZHEIMER’S DISEASE Article 28 September 2022 SINGLE-CELL EPIGENOMIC ANALYSES IMPLICATE CANDIDATE CAUSAL VARIANTS AT INHERITED RISK LOCI FOR
ALZHEIMER’S AND PARKINSON’S DISEASES Article 26 October 2020 AN INTEGRATED MULTI-OMICS APPROACH IDENTIFIES EPIGENETIC ALTERATIONS ASSOCIATED WITH ALZHEIMER’S DISEASE Article 28 September
2020 BACKGROUND & SUMMARY Alzheimer’s disease (AD) is a prevalent neurodegenerative disorder among the aged population. The main clinical features include memory and learning deficits,
disorientation, mood swings, and behavioral issues1. Studies of patients with familial (early-onset) AD identified autosomal dominant mutation of the amyloid precursor protein (_APP_),
presenilin 1, and presenilin 22. However, these mutations account for only 1%–5% of the total disease burden3. Most cases of AD are late-onset (>65 years), which are caused by complex
crosstalk of genetic and environmental factors4,5. Genome-wide association studies have identified many risk genes6,7,8,9. These genes function in diverse biological processes, such as
immune system process (_TNF_, _IL8_, _CR1_, _CLU_, _CCR2_, _PICALM_, and _CHRNB2_), cellular membrane organization (_SORL1_, _APOE_, _PICALM_, _BIN1_, and _LDLR_), and endocytosis (_PICALM_,
_BIN1_, and _CD2AP_)10. However, the identified AD risk-associated genes only contribute to a small portion of AD pathogenesis11, thus limiting their application in causal mechanism studies
and new drug discovery12. For sporadic AD, age is the biggest risk factor for AD genesis13. Studies suggest that AD and aging are intrinsically interwoven with each other14,15. For example,
brains of elder individuals contain abnormal deposits of aggregated proteins such as hyperphosphorylated tau (p-tau), amyloid-_β_ (A _β_), and _α_-synuclein16; however, it remains unclear
whether they are linked to AD genesis. For AD studies, an open question is if there is any molecular mechanism, especially aging-related mechanism, mediating these diverse biological
processes. Recently, an increasing number of studies have revealed the importance of the dysregulation occurring in cis- or trans-regulatory regions. Expression quantitative trait loci
(eQTL) analysis supports the proposition that AD risk-associated single nucleotide polymorphisms (SNPs) take regulatory roles by affecting the expression of nearby AD genes in the form of
looped interactions6,17,18,19. Large-scale DNA methylation studies have identified hypo- and hyper-methylated enhancers in postmortem AD brain samples, which alter the regulation of AD risk
genes20,21,22. H3K27ac, a marker for active enhancers and promoters, is differentially distributed at the regulatory regions involved in the progression of amyloid-_β_ and tau pathology23.
Epigenetic studies on other histone marks, for example, H3K9me324, H3K9ac25, and H4K16ac26, have also revealed critical links from epigenomic dysregulation to AD genesis. Meanwhile, studies
of epigenetic drugs have suggested the benefits of epigenetic modification. For example, inhibition of the HDAC3 protein by RGFP966 can reverse AD-related pathologies _in vitro_ and _in
vivo_ mouse models27. In our previous study using large-scale AD patients, we predicted that AD patients suffer from transcription regulation degeneration, which disrupts many AD-related
pathways28. However, it is still not clear how the changes in the non-coding regulatory regions, especially epigenetic changes, contribute to AD genesis. It requires a detailed map of
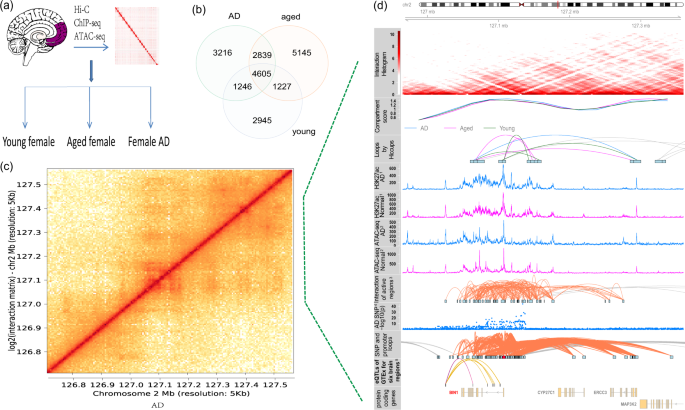
long-distance interaction to link regulatory regions to disease genes29. In this dataset, we generated high-resolution maps of three-dimensional (3D) chromatin architecture of aging and AD
using Hi-C technology. The prefrontal cortex region of post-mortem brain tissue from dementia-free elderly females (hereafter called “aged”, n = 4, mean age = 90, Chinese), female patients
with AD (n = 4, mean age = 91.5, Chinese), and cognitively normal young females (hereafter called “young”,n = 3, mean age = 29, Chinese) were used for Hi-C sequencing (see Fig. 1a and Table
1). All the selected AD samples were carefully evaluated so that AD patients all had similar and severe disease conditions; aged and young samples were free of dementia. During the
sequencing step, two samples were randomly selected from the aged and AD groups to generate 800 million paired-end reads with an estimated resolution of 9000 bp. The other samples were
sequenced for 3 billion reads with an estimated resolution of 3000 bp. After merging Hi-C data from the same group, the HiCCUPS tool identified 11906, 13816, and 10023 loops at a cutoff of
FDR <0.1 for AD, aged, and young groups, respectively (see Fig. 1b). To perform integrative analysis, other data were also generated or collected to facilitate the understanding of Hi-C
results, e.g., ATAC-seq, H3K27ac ChIP-seq, and GWAS SNPs. As an example, Fig. 1c,d shows the integrated results for _BIN1_ gene and the surrounding regions. Compared with nearby genes, more
Hi-C loops, H3K27ac marks, ATAC-seq signals, and AD GWAS SNPs are observed from upstream to gene body of _BIN1_, indicating that _BIN1_ is under intensive regulation. The Hi-C analysis
results are also presented, including interactions of active regulatory regions and SNP-promoter interactions. This result indicates that long-distance interactions are closely related to
_BIN1_ activities. The analysis results for more genes and genomic regions are available in http://menglab.pub/hic. METHODS SAMPLE COLLECTION FOR HI-C STUDY Postmortem brain samples
(prefrontal cortex regions) of 11 female individuals, including 4 subjects diagnosed with AD (Braak NFT stage >4, mean age = 91.5), 4 age-matched normal subjects (mean age = 90), and 3
young subjects (mean age = 29), were collected from the Chinese Brain Bank Center in Wuhan (CBBC, http://cbbc.scuec.edu.cn) and China Brain Bank, Zhejiang University
(http://www.neuroscience.zju.edu.cn). Informed consent for autopsy had been provided by all subjects during life. This study was reviewed and approved by the Ethics Committee of both brain
banks and Shanghai University of Chinese Medicine. The clinical information of each subject was reviewed by independent neurologists with expertise in dementia, and the neuropathological
diagnosis was given regarding the most likely clinical diagnosis at the time of death. AD samples were carefully evaluated so that all included AD subjects with homogeneous disease status.
The following criteria are more considered: (1) Braak NFT stage of ≥5 and severe disease stages; (2) within an age range of 85 to 95; (3) no or weak neuronal loss; and (4) not affected by
other neurological diseases. HI-C The AD, aged, and young samples were randomly labeled, and the sample information was blind to experimental staff. The Hi-C experiment was performed
following the protocol introduced in30. Brain cells were suspended in lysis buffer (10 mMTris, pH 8.0, 10 mM NaCl, 0.2% Igepal CA-630 and 1 × cOmplete_TM_ protease inhibitors (Sigma-Aldrich,
11697498001) and incubated on ice for 10 mins. Centrifugation at 2500 g for 5 mins at 4 °C, followed by removal of supernatant. Resuspended in 342 _μ_L 1 × NEBuffer 3.1, and incubated with
38 _μ_L 10% SDS at 65 °C for 10 mins. Added 43 _μ_L of 10% Triton X-100 to the Hi-C-tube to quench the SDS at 37 pellet for 15 min. Added 12 _μ_L 10 × NEBuffer 3.1 and 400U DpnII (NEB,
R0543), and mixed to digest the chromatin overnight at 37pellet on a rocking platform. Inactivated DpnII restriction enzyme at 65 °C for 25 mins. Then, biotin-14-dATP (Life Technologies,
19524-016), dCTP, dGTP, dTTP and DNA polymerase I Kenow were added (NEB, M0210), and incubated at 23 °C for 4 h. The digested chromatin was diluted and re-ligated by T4 DNA ligase (NEB,
M0202), incubated at 16 °C for 4 h, and shaken for three times. De-cross-linked by adding 30 _μ_L proteinase K, and incubated at 65 °C overnight. The DNAs were extracted and dissolved in 50
_μ_l 10 mMTris, pH 8.0. Then T4 DNA polymerase (NEB, M0203) was added and removed biotin for 4 hr at 20 °C, and the enzymes were inactivated for 20 mins at 75 °C. The DNAs were sheared to a
size of 300 bp using Covaris M220. Pulldown with Streptavidin T1 beads (Life Technologies, 65602). Then, performed end repair, A adding, adaptor adding reaction, PCR amplification and DNA
products size selection. The libraries were sequenced by the Illumina NovaSeq. 6000 sequencing platform. HI-C DATA ANALYSIS The raw sequencing data were cleaned with the trimmomatic tool
under the default setting31. The cleaned fastq data were input to HiC-Pro pipeline32 to generate non-duplicated valid pairs, and we recorded the genomic interactions reported by ligated
reads. The UCSC hg38 genome was used for alignments. The quality of analysis results in each step was evaluated following the protocol introduced in https://www.encodeproject.org/pipelines/,
including inter-/intra-chromosomal pairs, chimeric pairs, duplicates, intra-fragment, intra-long distance ranges, and ligations. The sparse interaction matrices were generated at different
bin sizes, ranging from 2000 to 200,000 bp. The compartment discovery and differential compartment activity analysis were performed using HOMER33 with a bin size of 25,000 bp under the
default parameter setting. The first principal component (PC1) of principal component analysis (PCA) was used to indicate compartment A/B along the genome. During this step, the samples from
paired groups were input for differential compartment activity analysis. To avoid the flipped signs of PC1 values, we applied two steps: (1) we compared the signs of each bin across the
samples of the same groups; (2) we used H3K27ac signals to decide PC1 signs of bins. The PC1 value along the hg38 genome was recorded in bedGraph format for visualization. LOOPS DISCOVERY
ANALYSIS The resolution of Hi-C data of each sample was estimated by applying juicer tool. We found that the resolution of 9 samples with higher sequencing depth was about 3200 bp and two
samples had a resolution of 8500 bp. The loops were predicted using HiCCUPS34 at a cutoff of FDR <0.1 at two bin sizes of 5000 and 10000 bp, respectively. To further improve the
resolution, the valid pairs generated by HiC-Pro tool from the same sample group were merged together and then transformed into *.hic files. The loops were predicted using HiCCUPS under
default parameter settings. By analyzing the HiCCUPS output, many loops were reported in only one or two groups. We checked the contacting frequency for these group-specific loops in other
groups and did not find any loop with completely loop loss or gain in all three groups. Therefore, the loops identified in AD, aged, and young samples were merged into non-overlapped ones.
In this process, bedtools35 was used by setting the minimum overlap as 5000 bp or the max length of loop anchors, and ensuring that there was zero gap. The self-contacted loops were filtered
so that the anchoring regions of the same loops were not overlapped36. TADS DISCOVERY ANALYSIS Topologically associating domains (TAD) were discovered with HOMER using the script of
findTADsAndLoops.pl33. This tool works by generating relative contact matrices for each chromosome and scanning them for locally dense regions of triangle domains that have a high degree of
inter-domain interactions relative to their surrounding region. In this step, we set the resolution to 3000 bp and an overlapping window size of 15000 to find the TADs. NORMALIZED CONTACT
MATRICES The raw contact matrices were generated by Hic-Pro at an arbitrary bin size of 5000 bp or 10000 bp. To generate comparable contact matrices, the raw interaction matrices of 11
samples were normalized using the R tool multiHiCcompare37. We firstly filtered the interactions with a total frequency of less than 20. In this step, only the intra-chromosomal interaction
was considered. Under the default parameter setting, a normalized contact matrix for each chromosome was generated under default parameter setting. To evaluate the quality of Hi-C data, we
performed clustering analysis, including principal component analysis and hierarchical clustering, using all whole interaction profiles. Our analysis found that sequencing depth or ratio of
uniquely mapped reads had a significant impact on the output matrices. Similar results were also observed with the reproducibility analysis using the raw matrix data38. Therefore, the
normalized interaction matrices were adjusted to remove their effects using the ComBat tool in R package sva39. In this process, 11 samples were classified and labelled as high-resolution
and medium-resolution samples; the ratios of uniquely mapped reads were treated as a continuous covariate. MAPPING REGIONS TO THE GENE BODY The anchor regions of loops or differential
interactions were mapped to the gene bodies by R package ChIPseeker40. The gene body annotation was based on known genes from UCSC build hg38, including promoter, 5′UTR, 3′UTR, exon, intron,
downstream, and intergenic regions. The promoter regions were defined as regions from −2000 bp to 2000 bp around TSS. Most human genes have multiple promoters, and these promoters were all
considered. ASSAY FOR TRANSPOSASE-ACCESSIBLE CHROMATIN USING SEQUENCING (ATAC-SEQ) ATAC-seq was performed in GENEWIZ company following the protocol introduced in41,42. Postmortem brain
samples in the prefrontal cortex regions of 26 individuals, including 13 diagnosed with AD and 13 normal subjects were collected from the Chinese Brain Bank Center in Wuhan (CBBC,
http://cbbc.scuec.edu.cn) and China Brain Bank, Zhejiang University (http://www.neuroscience.zju.edu.cn). Then, place frozen tissue into a pre-chilled 2 ml Dounce with 2 ml cold nuclei lysis
buffer. Allow frozen tissue to thaw for 5 minutes. Dounce with A pestle until resistance goes away (10 strokes). Dounce with B pestle for 20 strokes. Pre-clear larger chunks by pelleting at
100 RCF for 1 min in a pre-chilled centrifuge.Count nuclei using Trypan blue staining and aliquot nuclei for ATAC reaction. Harvest and count cells. Cells should be intact and in a
homogenous, single-cell suspension; Centrifuge 50,000 cells 5 min at 500 × g, 4 °C. The number of cells at this step is crucial, as the transposase-to-cell ratio determines the distribution
of DNA fragments generated. Remove and discard supernatant. Wash cells once with 50 _μ_l of cold PBS buffer. Centrifuge 5 min at 500x g, 4 °C. Remove and discard supernatant. Gently pipet up
and down to resuspend the cell pellet in 50 _μ_l of cold lysis buffer. Centrifuge immediately for 10 min at 500 × g, 44 °C. Discard the supernatant, and immediately continue to
transposition reaction. Make sure the cell pellet is set on ice. To make the transposition reaction mix, combine the following: TD (2x reaction buffer from Nextera kit) 25 _μ_l; TDE1
(Nextera Tn5 Transposase from Nextera kit) 2.5 _μ_l; Nuclease-free H2O 22.5 _μ_l. Resuspend nuclei pellet in the transposition reaction mix. Incubate the transposition reaction at 37 °C for
30 min. Gentle mixing may increase fragment yield. Immediately following transposition, purify using a Qiagen MinElute PCR Purification Kit. Elute transposed DNA in 10 _μ_l Elution Buffer
(Buffer EB from the MinElute kit consisting of 10 mM Tris·Cl, pH 8). To amplify transposed DNA fragments, combine the transposed DNA (10 _μ_l), nuclease-free H2O (10 _μ_l),25 _μ_M PCR Primer
1 (2.5 _μ_l), 25 _μ_M Barcoded PCR Primer 2 (2.5 _μ_l), NEBNext High-Fidelity 2x PCR Master Mix (2 5 _μ_l). Thermal cycle as follows 72 °C,5 min, 1 cycle; 98 °C,30 sec; 98 °C, 10 sec 5
cycles; 63 °C, 30 sec; 2 °C, 2 min; 4 °C. CIS-REGULATORY REGIONS In this work, we used H3K27ac and ATAC-seq signals to define cis-regulatory regions (CREs). Raw data of H3K27ac ChIP-seq were
collected from the GEO database with ID of GSE10253823, where 47 post-mortem entorhinal cortex tissue samples were used to identify widespread AD-associated acetylomic variations. Cleaned
fastq files were aligned to the human genome hg38 following the instructions of the original paper. In this step, duplicated reads were removed. The sorted and indexed bam files were merged
together by samtools43 into a single bam file, and then we performed peak calling using macs2 under a parameter setting of “–keep-dup all–broad–broad-cutoff 0.1”. ATAC-seq data were
generated and analyzed as described in our previous work28, including the prefrontal cortex regions of 13 Chinese people with diagnosed with AD and 13 Chinese normal subjects. Like ChIP-seq
data, AD and normal samples were merged for peak calling with a parameter setting of “–keep-dup all–nomodel–shift −100–extsize 200”. We applied bedtools to check the peak overlaps of H3K27ac
ChIP-seq and ATAC-seq. The peaks for H3K27ac marks and ATAC-seq were merged into non-overlapped regions, which were treated as active regions of the brain. We mapped these regions to gene
bodies and classified them as promoters, enhancers, and other regions. The active regions that locate in the regulatory region, are treated as CREs. AD RISK SNPS The AD GWAS analysis results
were collected from a recently published meta-analysis on PGC-ALZ, IGAP, ADSP, and UKB6,7,8,9. The SNPs and their significance were downloaded from
https://ctg.cncr.nl/software/summary_statistics. The AD risk SNPs were selected with a cutoff of _p_ < 1_e_-5, and their genomic locations were transformed into the corresponding
locations on hg38 genome. In total, 6468 SNPs were selected to check their overlap with Hi-C interactions and active regions or enhancers. HI-C LOOPS OF SNP-PROMOTER INTERACTIONS The genomic
locations of AD risk SNPs were mapped to the human genome based on the annotation of dbSNP database. We filtered the SNPs located in the promoter regions (±2000 bp around TSS). The TSS
information was collected from the ENSEMBL database and the promoter regions were defined as from −2000bp to 2000 bp around TSS. Most human genes have multiple promoters, and these promoters
were all considered. Next, we identified the loops linking SNPs to promoters. It is known that the functional SNPs in the noncoding regions usually take roles by affecting the transcription
factor binding. However, there is no golden standard to define the ranges of cis-regulatory regions. Here, we arbitrarily set a region of 1000 bp around SNPs as the SNP-affected
cis-regulatory regions. The contacting frequency between promoter and SNP regions was calculated by _bedtools findoverlaps_, which counted the number of reads that anchored at both SNP and
promoter regions. To find a proper cutoff of contacting frequency, we calculated the frequencies of all possible SNP-promoter pairs on the same chromosome and we found that most of the pairs
have a contact frequency of 0. Among the pairs linked by at least one Hi-C read, there are less than 1% of SNP-promoter pairs with frequency >20 (_p_ < 0.01). If the contacting
frequency was great than 20, the corresponding SNP-promoter regions were supposed to be linked by loops. Additionally, we also performed loop discovery using bin-free tool Binless44 under
the suggested parameter setting. For interaction profiles of active regions, we applied R package InteractionSet to calculate the interaction frequency between active regulatory regions. The
pairs with less than 20 interactions or spanning different chromosomes were filtered. EQTL FOR HUMAN BRAIN REGIONS The significant eQTLs were downloaded from the GTEx database
(https://gtexportal.org/), where eQTLs had been filtered at a cutoff of FDR <0.05. Based on text mining, we selected eQTLs of six brain regions: the amygdala, anterior cingulate cortex,
cortex, frontal cortex, hippocampus, and hypothalamus. As an independent validation, we also included the eQTLs stored in BRAINEAC database (http://www.braineac.org/)45 to evaluate the
interactions reported by GTEx or Hi-C datasets. MSBB DATA ANALYSIS The RNA-seq and clinical data of AD patients were downloaded from the AMP-AD project
https://www.synapse.org/#!Synapse:syn2580853. To simplify the analysis, only MSBB data were selected. After filtering the sample with incomplete information, MSBB data included 223 AD
patients and four brain regions: the frontal pole (BA10), superior temporal gyrus (BA22), parahippocampal gyrus (BA36), and frontal cortex (BA44). These subjects had diverse clinical
manifestations, for example, cognitive score and Braak stages. Approximately 61% were diagnosed as having pathological AD or probable AD. The clinical dementia rating scale (CDR) and
mini–mental state examination (MMSE) severity tests were used to assess cognitive status. Based on CDR classification, subjects were grouped as no cognitive deficits (CDR = 0), questionable
dementia (CDR = 0.5), mild dementia (CDR = 1.0), moderate dementia (CDR = 2.0), and severe to terminal dementia (CDR = 3.0–5.0). The differential expressed genes were identified by the R
limma package, where two conditions were compared for all the expressed genes. VISUALIZATION OF LONG-DISTANCE INTERACTION The interaction matrices for AD, aged, and young groups were
generated by merging the matrices from different samples. Then the interaction matrices were normalized by the Knight-Ruiz (KR) method37. An interaction heatmap was generated by
HiCPlotter46. R package Gviz was used to visualize the binned triangle interaction heatmap, compartment A/B, H3K27ac ChIP-seq, ATAC-seq, GWAS SNP significance signal, and mapped genes along
the human genome. The contacting loops were mapped by R package GenomicInteractions47. DATA RECORDS The Hi-C raw fastq files were deposited at NCBI under accession number of SRP28018348. The
raw ATAC-seq data were publicly available in the Gene Expression Omnibus (GEO) database with the ID of GSE12904149. To facilitate the usage of Hi-C datasets, we build a shiny-based tool at
http://menglab.pub/hic/. It can perform integrative analysis for genes or user-defined genomic regions, including an interaction histogram, compartment score, loops predicted by HiCCUPS,
H3K27ac peaks, open chromatin regions by ATAC-seq, contacts of active regions, AD risk SNPs, SNP-promoter interactions, eQTLs, and protein-coding genes. The processed Hi-C data are also
available for public download in the same web tool. TECHNICAL VALIDATION QUALITY ASSESSMENT OF HI-C DATA Quality assessment of each Hi-C dataset was performed following the protocol
introduced in https://www.encodeproject.org/pipelines/, including inter-/intra-chromosomal pairs, chimeric pairs, duplicates, intra-fragment, intra long distance ranges, and ligations (see
here50,51). Figure 2a shows the results of reads mapping step using one example of AD samples. >90% of reads were aligned on the genome of hg38 by bowtie252. Figure 2b shows the pairing
statistics by HiC-Pro, in which more than 60% reads were paired. Among the pairs, more than 70% are validated pairs (see Fig. 2c). Among the validated pairs, there are about 30% of
duplicates, which is mainly due to the high sequencing depth (see Fig. 2d). Fragment size distribution was extracted from the valid interaction and we observed a distribution centered around
300 bp, which corresponds to the paired-end insert size commonly used (see Fig. 2e). For the other samples, we also perform the same quality assessment and observed similar results50. We
also performed reproducibility analysis by HICRep tool38 among samples. We found that the samples had overall good similarity (>0.85) in interaction profiles (see Fig. 2f). Overall, all
the evaluation results suggested a good quality of the Hi-C data. HI-C INTERACTION SITES OVERLAP WITH PROMOTERS AND ENHANCERS For the Hi-C loops, we found that 60% of them were overlapped by
both H3k27ac and ATAC-seq peaks (see Fig. 3a). We also checked the genomic distribution of anchor sites of Hi-C loops and found that more than 40% of the loops overlapped with gene promoter
regions within 1000 bp around transcription start sites (TSS). Moreover, 18% of the loops were mapped to the distal intergenic region50. Compartment A/B analysis indicated that the TSS
regions were more active in compartment activity (see Fig. 3b). All of these results suggested that Hi-C loops were more related to active regulatory regions on the chromatins. We
investigated the relationship among long-distance interaction, chromatin status, and gene expression. Hence, we checked whether long-distance interactions contributed to gene expression
regulation using a method introduced in53. We identified 2704 differentially expressed genes (DEGs) in AD patients using the MSBB dataset from AMP-AD projects. We found that fold changes of
DEGs had a good correlation with the changes in long-distance interactions, and the Spearman’s correlation was 0.3 (_p_ = 2.36_e_-57, see Fig. 3c), which is comparable to findings in neuron
cells53,54. LINK AD RISK SNPS TO RISK GENES To build the links from AD risk SNPs to genes, 6468 AD risk SNPs were collected from published GWAS data6 at a cutoff of _p_ < 1.0_e_-5. After
filtering, 3498 SNPs located within promoter regions were selected for SNP-promoter loop discovery. We identified 75,953 SNP-promoter links with a contacting frequency of >20. They
included 2771 AD risk SNPs and 355 genes55. We attempted to evaluate if existing brain eQTL can help to link AD-related enhancers to their target genes. Therefore, we collected 7561
significant eQTLs from the GTEx database for six brain regions (the amygdala, anterior cingulate cortex, cortex, frontal cortex, hippocampus, and hypothalamus). Among these eQTLs, 3417 were
overlapped with Hi-C loops. Figure 4a shows the number of eQTLs and their overlaps with Hi-C loops in the six brain regions. Overall, a modest overlap was observed between Hi-C loops and
eQTL. SNP-promoter links were then evaluated using the top 10 AD risk genes in the AlzGen database (see Fig. 4b). The promoters of nine genes were heavily contacted with the AD-associated
SNPs. However, eQTLs only reported the links for one gene, _CR1_; that is, eQTL almost failed to identify the target genes of AD-associated SNPs. Taking _BIN1_ as an example, there were 126
AD-associated SNPs along the gene body and upstream regions. Hi-C data supported that 63 of them were linked to the promoter of the _BIN1_ gene(see Fig. 4c), which validated the roles of
AD-associated SNPs in the activity of _BIN1_. Among AD-associated SNPs within or surrounding the _BIN1_ gene, rs4663105 was the most associated SNP, with a significance of _p_ = 1.45_e_-44.
However, it had no link with the _BIN1_ promoter. Similar results were observed for other AD risk SNPs, including the six most associated SNPs. Among the SNPs with Hi-C links to _BIN1_,
rs35103166, an SNP located in the upstream region of _BIN1_, was associated with AD at _p_ = 4.79_e_-23. The contacts between rs35103166 and _BIN1_ promoters were supported by 99 reads,
supporting a high-confidence interaction. Similar results were observed with other AD risk SNPs. Unlike with Hi-C loops, we did not find any eQTL link between AD-associated SNPs and _BIN1_
expression. We also checked the eQTLs reported by BRAINEAC database45 and did not find eQTL links to _BIN1_ under a cutoff of _p_ < 0.01. Another example can be seen with _CR1_ (see Fig.
4d), which was linked by eQTLs of multiple AD-associated SNPs. There were 48 AD-associated SNPs near or within the _CR1_ gene, among which 37 SNPs were linked to _CR1_ by 67 eQTLs in the six
brain regions. Most of these SNP-gene links were supported by Hi-C SNP-promoter loops. Hi-C loops reported more SNP-promoter links for 43 AD risk SNPs. INTEGRATING HYPER-ACETYLATED PEAKS TO
IDENTIFY THE DYSREGULATED ENHANCER-PROMOTER INTERACTIONS The expensive cost of Hi-C experiments limits its application to too many samples. To identify the changed enhancer-promoter
regulation, a feasible solution is to integrate Hi-C annotation with other dysregulated signals. As a demonstration, we collected 1475 hyper-acetylated H3K27ac peaks from the published
study23 and identify the target genes of 806 peaks56. Figure 5a shows the results of a peak on chr13:112101248–112102698, which was reported as the most hyper-acetylated peak. This peak
located at the downstream of _ARHGEF7_ and _TEX29_ genes. It interacted with the promoter of ENST00000483189, one transcript of _ARHGEF7_ gene. This result suggested that _ARHGEF7_ might
take a more important role in AD. It should be noticed that _ARHGEF7_ was not the proximal gene according to their genomic location, which suggested a necessity to use Hi-C results to
annotate dysregulated regulatory elements. Figure 5b shows another example of the most hyper-acetylated peak on chr5:640598–642071, located in the ninth intron of _CEP72_. It interacted with
the promoter of ENST00000512038, a transcript of _CEP72_. Under our parameter setting, we failed to identify links from some top-ranked hyper-acetylated peaks and it is not clear if they
are involved in transcription regulation. One of the hyper-acetylated peaks of chr17:43959954–43961546 is proximal to the _MAPT_ gene. Figure 5c shows its chromatin interaction with _MAPT_
transcript promoters. Multiple _MAPT_ transcripts were linked to this peak, e.g. ENST00000571311, ENST00000420682, and ENST00000262410. Additionally, the same peak is also linked to the
promoters of multiple transcripts of _CRHR1_ gene. _CRHR1_ has been reported for association with synaptic loss and memory in other neurological diseases57. This result might indicate its
potential involvement in AD. CODE AVAILABILITY The Hi-C data analyses were performed using public tools. The following softwares were used to perform Hi-C data analysis: 1. FastQC v0.11.9
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ 2. HiC-Pro v2.11.3 https://github.com/nservant/HiC-Pro 3. Juicer tools v1.14.08 https://github.com/aidenlab/Juicebox 4. HOMER V2.0
http://homer.ucsd.edu/homer/interactions2/index.html 5. bedtools v2.29.2 https://bedtools.readthedocs.io/en/latest/ 6. Gviz V1.40.0
https://bioconductor.org/packages/release/bioc/html/Gviz.html 7. ChIPseeker v1.32.0 http://bioconductor.org/packages/release/bioc/vignettes/ChIPseeker 8. HiCPlotter v0.6.6
https://github.com/akdemirlab/HiCPlotter 9. multiHiCcompare v1.14.0 https://dozmorovlab.github.io/multiHiCcompare/ 10. MACS v2.2.6 https://github.com/macs3-project/MACS 11. R v3.6.2
https://cran.r-project.org/ 12. limma v3.1.2 https://bioconductor.org/packages/release/bioc/html/limma.html REFERENCES * Bateman, R. J. _et al_. Clinical and biomarker changes in dominantly
inherited alzheimer’s disease. _N Engl J Med_ 367, 795–804, https://doi.org/10.1056/NEJMoa1202753 (2012). Article CAS Google Scholar * Bekris, L. M., Yu, C.-E., Bird, T. D. & Tsuang,
D. W. Review article: Genetics of alzheimer disease. _Journal of Geriatric Psychiatry and Neurology_ 23, 213–227, https://doi.org/10.1177/0891988710383571 (2010). Article Google Scholar *
Garre-Olmo, J. Epidemiology of alzheimer’s disease and other dementias. _Revista de neurologia_ 66, 377–386, https://doi.org/10.12688/f1000research.50786.1 (2018). Article CAS Google
Scholar * Dorszewska, J., Prendecki, M., Oczkowska, A., Dezor, M. & Kozubski, W. Molecular basis of familial and sporadic alzheimer’s disease. _Current Alzheimer Research_ 13, 952–963,
https://doi.org/10.2174/1567205013666160314150501 (2016). Article CAS Google Scholar * Bellenguez, C., Grenier-Boley, B. & Lambert, J.-C. Genetics of alzheimer’s disease: where we
are, and where we are going. _Current Opinion in Neurobiology_ 61, 40–48, https://doi.org/10.1016/j.conb.2019.11.024 (2020). Article CAS Google Scholar * Jansen, I. E. _et al_.
Genome-wide meta-analysis identifies new loci and functional pathways influencing alzheimer’s disease risk. _Nature genetics_ 51, 404–413, https://doi.org/10.1038/s41588-018-0311-9 (2019).
Article CAS Google Scholar * Kunkle, B. W. _et al_. Genetic meta-analysis of diagnosed alzheimer’s disease identifies new risk loci and implicates a _β_, tau, immunity and lipid
processing. _Nature Genetics_ 51, 414–430, https://doi.org/10.1038/s41588-019-0358-2 (2019). Article CAS Google Scholar * Schwartzentruber, J. _et al_. Genome-wide meta-analysis,
fine-mapping and integrative prioritization implicate new alzheimer’s disease risk genes. _Nature Genetics_ 53, 392–402, https://doi.org/10.1038/s41588-020-00776-w (2021). Article CAS
Google Scholar * Wightman, D. P. _et al_. A genome-wide association study with 1,126,563 individuals identifies new risk loci for alzheimer’s disease. _Nature Genetics_ 53, 1276–1282,
https://doi.org/10.1038/s41588-021-00921-z (2021). Article CAS Google Scholar * Olgiati, P., Politis, A. M., Papadimitriou, G. N., De Ronchi, D. & Serretti, A. Genetics of late-onset
alzheimer’s disease: update from the alzgene database and analysis of shared pathways. _International journal of Alzheimer’s disease_ 2011, https://doi.org/10.4061/2011/832379 (2011). *
Ebbert, M. T. _et al_. Population-based analysis of alzheimer’s disease risk alleles implicates genetic interactions. _Biological Psychiatry_ 75, 732–737,
https://doi.org/10.1016/j.biopsych.2013.07.008 (2014). Article CAS Google Scholar * Cummings, J., Feldman, H. H. & Scheltens, P. The “rights” of precision drug development for
alzheimer’s disease. _Alzheimer’s Research & Therapy_ 11, https://doi.org/10.1186/s13195-019-0529-5 (2019). * Price, J. L. _et al_. Neuropathology of nondemented aging: Presumptive
evidence for preclinical alzheimer disease. _Neurobiology of Aging_ 30, 1026–1036, https://doi.org/10.1016/j.neurobiolaging.2009.04.002 (2009). Article Google Scholar * Xia, X., Jiang, Q.,
McDermott, J. & Han, J.-D. J. Aging and alzheimer’s disease: Comparison and associations from molecular to system level. _Aging Cell_ 17, e12802, https://doi.org/10.1111/acel.12802
(2018). Article CAS Google Scholar * Meng, G., Zhong, X. & Mei, H. A systematic investigation into aging related genes in brain and their relationship with alzheimer’s disease. _PLOS
ONE_ 11, e0150624, https://doi.org/10.1371/journal.pone.0150624 (2016). Article CAS Google Scholar * Hou, Y. _et al_. Ageing as a risk factor for neurodegenerative disease. _Nature
Reviews Neurology_ 15, 565–581, https://doi.org/10.1038/s41582-019-0244-7 (2019). Article Google Scholar * Katsumata, Y., Nelson, P. T., Estus, S. & Fardo, D. W. Translating
alzheimer’s disease-associated polymorphisms into functional candidates: a survey of igap genes and snps. _Neurobiology of Aging_ 74, 135–146,
https://doi.org/10.1016/j.neurobiolaging.2018.10.017 (2019). Article CAS Google Scholar * Amlie-Wolf, A. _et al_. Inferring the molecular mechanisms of noncoding alzheimer’s
disease-associated genetic variants. _Journal of Alzheimer’s Disease_ 72, 301–318 (2019). Article CAS Google Scholar * Kikuchi, M. _et al_. Enhancer variants associated with alzheimer’s
disease affect gene expression via chromatin looping. _BMC Medical Genomics_ 12, https://doi.org/10.1186/s12920-019-0574-8 (2019). * De Jager, P. L. _et al_. Alzheimer’s disease: early
alterations in brain dna methylation at ank1, bin1, rhbdf2 and other loci. _Nature neuroscience_ 17, 1156–1163 (2014). Article Google Scholar * Li, P. _et al_. Epigenetic dysregulation of
enhancers in neurons is associated with alzheimer’s disease pathology and cognitive symptoms. _Nature communications_ 10, 1–14 (2019). ADS Google Scholar * Smith, R. G. _et al_. A
meta-analysis of epigenome-wide association studies in alzheimer’s disease highlights novel differentially methylated loci across cortex. _Nature Communications_ 12,
https://doi.org/10.1038/s41467-021-23243-4 (2021). * Marzi, S. J. _et al_. A histone acetylome-wide association study of alzheimer’s disease identifies disease-associated h3k27ac differences
in the entorhinal cortex. _Nature neuroscience_ 21, 1618–1627 (2018). Article CAS Google Scholar * Lee, M. Y. _et al_. Epigenome signatures landscaped by histone h3k9me3 are associated
with the synaptic dysfunction in alzheimer’s disease. _Aging Cell_ 19, https://doi.org/10.1111/acel.13153 (2020). * Klein, H.-U. _et al_. Epigenome-wide study uncovers large-scale changes in
histone acetylation driven by tau pathology in aging and alzheimer’s human brains. _Nature Neuroscience_ 22, 37–46, https://doi.org/10.1038/s41593-018-0291-1 (2018). Article CAS Google
Scholar * Nativio, R. _et al_. Dysregulation of the epigenetic landscape of normal aging in alzheimer’s disease. _Nature neuroscience_ 21, 497–505 (2018). Article CAS Google Scholar *
Janczura, K. J. _et al_. Inhibition of HDAC3 reverses alzheimer’s disease-related pathologies _in vitro_ and in the 3xtg-AD mouse model. _Proceedings of the National Academy of Sciences_
115, E11148–E11157, https://doi.org/10.1073/pnas.1805436115 (2018). Article CAS Google Scholar * Meng, G. _et al_. Accumulated degeneration of transcriptional regulation contributes to
disease development and detrimental clinical outcomes of alzheimer’s disease. _biorxiv_ https://doi.org/10.1101/779249 (2019). Article Google Scholar * Yu, J., Hu, M. & Li, C. Joint
analyses of multi-tissue hi-c and eQTL data demonstrate close spatial proximity between eQTLs and their target genes. _BMC Genetics_ 20, https://doi.org/10.1186/s12863-019-0744-x (2019). *
Belaghzal, H., Dekker, J. & Gibcus, J. H. Hi-c 2.0: An optimized hi-c procedure for high-resolution genome-wide mapping of chromosome conformation. _Methods_ 123, 56–65 (2017). Article
CAS Google Scholar * Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for illumina sequence data. _Bioinformatics_ 30, 2114–2120,
https://doi.org/10.1093/bioinformatics/btu170 (2014). Article CAS Google Scholar * Servant, N. _et al_. Hic-pro: an optimized and flexible pipeline for hi-c data processing. _Genome
biology_ 16, 259 (2015). Article Google Scholar * Heinz, S. _et al_. Transcription elongation can affect genome 3d structure. _Cell_ 174, 1522–1536.e22,
https://doi.org/10.1016/j.cell.2018.07.047 (2018). Article CAS Google Scholar * Durand, N. C. _et al_. Juicer provides a one-click system for analyzing loop-resolution hi-c experiments.
_Cell Systems_ 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016). Article CAS Google Scholar * Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for
comparing genomic features. _Bioinformatics_ 26, 841–842, https://doi.org/10.1093/bioinformatics/btq033 (2010). Article CAS Google Scholar * Meng, G. _et al_. TSD: A Computational Tool To
Study the Complex Structural Variants Using PacBio Targeted Sequencing Data. _G3 Genes|Genomes|Genetics_ 9, 1371–1376, https://doi.org/10.1534/g3.118.200900 (2019). Article CAS Google
Scholar * Stansfield, J. C., Cresswell, K. G. & Dozmorov, M. G. multiHiCcompare: joint normalization and comparative analysis of complex hi-c experiments. _Bioinformatics_ 35,
2916–2923, https://doi.org/10.1093/bioinformatics/btz048 (2019). Article CAS Google Scholar * Yang, T. _et al_. HiCRep: assessing the reproducibility of hi-c data using a stratum-adjusted
correlation coefficient. _Genome Research_ 27, 1939–1949, https://doi.org/10.1101/gr.220640.117 (2017). Article CAS Google Scholar * Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A.
E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. _Bioinformatics_ 28, 882–883,
https://doi.org/10.1093/bioinformatics/bts034 (2012). Article CAS Google Scholar * Yu, G., Wang, L.-G. & He, Q.-Y. ChIPseeker: an r/bioconductor package for ChIP peak annotation,
comparison and visualization. _Bioinformatics_ 31, 2382–2383, https://doi.org/10.1093/bioinformatics/btv145 (2015). Article CAS Google Scholar * Corces, M. R. _et al_. An improved
atac-seq protocol reduces background and enables interrogation of frozen tissues. _Nature methods_ 14, 959 (2017). Article CAS Google Scholar * Buenrostro, J. D., Wu, B., Chang, H. Y.
& Greenleaf, W. J. Atac-seq: a method for assaying chromatin accessibility genome-wide. _Current protocols in molecular biology_ 109, 21–29 (2015). Article Google Scholar * Li, H. _et
al_. The sequence alignment/map format and SAMtools. _Bioinformatics_ 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009). Article CAS Google Scholar * Spill, Y. G.,
Castillo, D., Vidal, E. & Marti-Renom, M. A. Binless normalization of hi-c data provides significant interaction and difference detection independent of resolution. _Nature
Communications_ 10, https://doi.org/10.1038/s41467-019-09907-2 (2019). * Ramasamy, A. _et al_. Genetic variability in the regulation of gene expression in ten regions of the human brain.
_Nature Neuroscience_ 17, 1418–1428, https://doi.org/10.1038/nn.3801 (2014). Article CAS Google Scholar * Akdemir, K. C. & Chin, L. HiCPlotter integrates genomic data with interaction
matrices. _Genome Biology_ 16, https://doi.org/10.1186/s13059-015-0767-1 (2015). * Harmston, N., Ing-Simmons, E., Perry, M., Barešić, A. & Lenhard, B. GenomicInteractions: An
r/bioconductor package for manipulating and investigating chromatin interaction data. _BMC Genomics_ 16, https://doi.org/10.1186/s12864-015-2140-x (2015). * Meng, G. Hi-c for ad. _NCBI
Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRP280183 (2020). * Meng, G. Transcriptional regulation loss disturbs the brain function and indicates detrimental clinical
outcomes of alzheimer’s disease. _GEO_ https://identifiers.org/geo/GSE129041 (2019). * Meng, G. Figure s1 the quality results reported by hic-pro pipeline. _figshare_
https://doi.org/10.6084/m9.figshare.21814986.v1 (2023). * Meng, G. Supplement_material_1 quality control of hi-c data. _figshare_ https://doi.org/10.6084/m9.figshare.21670412.v2 (2023). *
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with bowtie 2. _Nature Methods_ 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012). Article CAS Google Scholar *
Greenwald, W. W. _et al_. Subtle changes in chromatin loop contact propensity are associated with differential gene regulation and expression. _Nature communications_ 10, 1–17 (2019).
Article Google Scholar * Beagan, J. A. _et al_. Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression. _Nature Neuroscience_ 1–11 (2020). *
Meng, G. Table S1 Hyperhyperacetylated H3K27ac peaks and their target genes. _figshare_ https://doi.org/10.6084/m9.figshare.21815004.v1 (2023). * Meng, G. Table S2 The SNP-promoter
contacting information. _figshare_ https://doi.org/10.6084/m9.figshare.21815007.v1 (2023). * Cursano, S. _et al_. A CRHR1 antagonist prevents synaptic loss and memory deficits in a
trauma-induced delirium-like syndrome. _Molecular Psychiatry_ https://doi.org/10.1038/s41380-020-0659-y (2020). Article Google Scholar Download references ACKNOWLEDGEMENTS We are grateful
to the China Brain Bank of Zhejiang University School of Medicine and Chinese Brain Bank Center in Wuhan for providing human brain material. The results published here are in part based on
data obtained from the AMP-AD Knowledge Portal (https://doi.org/10.7303/syn2580853). We appreciate their generous contribution to the studies of Alzhiemer’s disease. We thank Dr. Lichun
Jiang and Dr. Ruping Sun for reading this manuscript and giving us many useful suggestions. The work was supported by National Natural Science Foundation of China(81973706, 81520108030,
21472238), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (16DZ2280200), the Scientific Foundation of Shanghai China (13401900103, 13401900101), the
National Key Research and Development Program of China (2019YFC1711000, 2017YFC1700200). AUTHOR INFORMATION Author notes * These authors contributed equally: Guofeng Meng, Hong Xu. AUTHORS
AND AFFILIATIONS * Institute of interdisciplinary integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China Guofeng Meng, Hong Xu, Dong Lu,
Shensuo Li, Zhenzhen Zhao & Weidong Zhang * Faculty of Business and Economics, The University of Hong Kong, Pokfulam Road, Hong Kong, China Haohao Li Authors * Guofeng Meng View author
publications You can also search for this author inPubMed Google Scholar * Hong Xu View author publications You can also search for this author inPubMed Google Scholar * Dong Lu View author
publications You can also search for this author inPubMed Google Scholar * Shensuo Li View author publications You can also search for this author inPubMed Google Scholar * Zhenzhen Zhao
View author publications You can also search for this author inPubMed Google Scholar * Haohao Li View author publications You can also search for this author inPubMed Google Scholar *
Weidong Zhang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS M.G. and Z.W. designed experiments. Z.W. coordinated sample collection. X.H.
and Z.Z. performed experiments. M.G., L.D. and L.S. performed the data analysis. L.H. performed statistical analysis. M.G. and Z.W. wrote the manuscript. CORRESPONDING AUTHORS Correspondence
to Guofeng Meng or Weidong Zhang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral
with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Meng, G., Xu, H., Lu, D. _et al._ Three-dimensional chromatin architecture datasets
for aging and Alzheimer’s disease. _Sci Data_ 10, 51 (2023). https://doi.org/10.1038/s41597-023-01948-z Download citation * Received: 06 July 2022 * Accepted: 10 January 2023 * Published:
24 January 2023 * DOI: https://doi.org/10.1038/s41597-023-01948-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Page not found - Eenadu.netContents of eenadu.net are copyright protected.Copy and/or reproduction and/or re-use of contents or any part thereof, w...
Benefit cap: estimated impact on parents, by age of youngest childOfficial Statistics BENEFIT CAP: ESTIMATED IMPACT ON PARENTS, BY AGE OF YOUNGEST CHILD Estimated impact of the benefit c...
Tribals become anthropologistsA woman in her mid-twenties, probably of European descent, is trying to explain something to a group of Bhil tribals in ...
Tania shergill expresses happiness over leading parade on republic day 2020Updated: Jan 24, 2020, 03:50 PM IST Captain Tania Shergill, first woman parade adjutant for the Republic Day parade expr...
Top six position just the start for beeston's women_ROD GILMOUR OF __THE HOCKEY PAPER__ SPEAKS TO BEESTON WOMEN, WHO ARE RIDING HIGH AHEAD OF THE FINAL MATCH BEFORE THE CH...
Latests News
Three-dimensional chromatin architecture datasets for aging and alzheimer’s diseaseABSTRACT Recently, increasing studies are indicating a close association between dysregulated enhancers and neurodegener...
10% reservation for general category: how political parties reacted4.Cong dubs govt's quota move as poll gimmick THE CONGRESS ON MONDAY SAID IT WILL SUPPORT A BILL TO GRANT 10 PER CE...
Locations | las cruces vet center | veterans affairsMAIN LOCATION 1120 Commerce Drive Suite B Las Cruces, NM 88011 * Mon. 8:00 a.m. to 4:30 p.m. * Tue. 8:00 a.m. to 4:30 p....
Idaho joins other states with informed consent for 'abortion reversal' proceduresSARAH MCCAMMON, HOST: Idaho has become the latest of a handful of states to pass a law promoting a medically-controversi...
Newell rubbermaid buys jarden, uniting two big consumer brandsNewell Rubbermaid said today that it will acquire Corp., paying nearly $16 billion to acquire all of its stock and debt....