Functional variants in a tttg microsatellite on 15q26. 1 cause familial nonautoimmune thyroid abnormalities
Functional variants in a tttg microsatellite on 15q26. 1 cause familial nonautoimmune thyroid abnormalities"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Insufficient thyroid hormone production in newborns is referred to as congenital hypothyroidism. Multinodular goiter (MNG), characterized by an enlarged thyroid gland with multiple
nodules, is usually seen in adults and is recognized as a separate disorder from congenital hypothyroidism. Here we performed a linkage analysis of a family with both nongoitrous congenital
hypothyroidism and MNG and identified a signal at 15q26.1. Follow-up analyses with whole-genome sequencing and genetic screening in congenital hypothyroidism and MNG cohorts showed that
changes in a noncoding TTTG microsatellite on 15q26.1 were frequently observed in congenital hypothyroidism (137 in 989) and MNG (3 in 33) compared with controls (3 in 38,722).
Characterization of the noncoding variants with epigenomic data and in vitro experiments suggested that the microsatellite is located in a thyroid-specific transcriptional repressor, and its
activity is disrupted by the variants. Collectively, we presented genetic evidence linking nongoitrous congenital hypothyroidism and MNG, providing unique insights into thyroid
abnormalities. SIMILAR CONTENT BEING VIEWED BY OTHERS THYROID HYPOGENESIS IS ASSOCIATED WITH A NOVEL _AKT3_ GERMLINE VARIANT THAT CAUSES MEGALENCEPHALY AND CORTICAL MALFORMATION Article Open
access 03 June 2022 GENOME-WIDE ASSOCIATION STUDY OF THYROID-STIMULATING HORMONE HIGHLIGHTS NEW GENES, PATHWAYS AND ASSOCIATIONS WITH THYROID DISEASE Article Open access 23 October 2023
CHROMOSOME 20P11.2 DELETIONS CAUSE CONGENITAL HYPERINSULINISM VIA THE LOSS OF _FOXA2_ OR ITS REGULATORY ELEMENTS Article Open access 11 April 2024 MAIN Thyroid hormones, triiodothyronine and
thyroxine (T4), regulate the metabolic activity of virtually all human tissues and cause, when depleted in children, growth restriction, intellectual disability and various symptoms. The
thyroid gland, an organ producing thyroid hormones, shows great size variation in humans from aplastic (<1 ml), normal (10–20 ml in adults) to enlarged (goiter; maximum >1,000 ml).
Congenital hypothyroidism occurs in about 1 in 2,000‒3,000 births worldwide1 and is recognized as one of the major preventable causes of intellectual disability. In developed countries,
universal newborn screening for congenital hypothyroidism was started in 1970s2, and most patients with congenital hypothyroidism have received early diagnosis and treatment. Congenital
hypothyroidism cases can be divided into the following two major categories: goitrous congenital hypothyroidism with an enlarged thyroid gland and nongoitrous congenital hypothyroidism with
a small- or normal-sized thyroid. From a clinical genetic standpoint, distinction between the two is important because goitrous congenital hypothyroidism is mostly due to autosomal recessive
Mendelian diseases such as the thyroglobulin defect3, the thyroid peroxidase defect4 and dual oxidase 2 defect5, whereas only small fraction (<10%) of nongoitrous congenital
hypothyroidism is explained by Mendelian diseases, including the thyroid-stimulating hormone (TSH) receptor defect and the PAX8 defect6,7. Five families presenting autosomal dominant
nongoitrous congenital hypothyroidism with a linkage region on the long arm of chromosome (chr) 15 were reported8, but the disease-causing variant(s) have not been clarified. Multinodular
goiter (MNG) is a disease characterized by the development of multiple thyroid nodules, which can be fluid-filled (cystic) or solid. Although MNG is usually benign, symptoms associated with
local compression may occur if left untreated. The exact cause of MNG is not well understood, although several risk factors such as age, female sex and iodine deficiency are known9. In this
study, we performed a linkage analysis of a Japanese family in which both nongoitrous congenital hypothyroidism and MNG were observed. Follow-up analyses with whole-genome sequencing (WGS)
of additional families and large-scale genetic screening led us to identify the nucleotide-level changes affecting a TTTG microsatellite that are responsible for the peculiar age-dependent
thyroid abnormalities. Marked expansions of microsatellite repeats have been known to cause neurodegenerative diseases such as Huntington’s disease10, but subtle changes in microsatellites
very rarely cause human diseases, including thyroid diseases. RESULTS LINKAGE ANALYSIS In the last 16 years, we have conducted targeted sequencing in 989 patients with congenital
hypothyroidism and determined the molecular diagnoses in 214 cases. Of the 282 cases with family history, only 63 were molecularly solved (Extended Data Fig. 1). Among the unsolved familial
cases, there was a large family (family A) with 13 individuals with nongoitrous congenital hypothyroidism, five with subclinical hypothyroidism and three with suspected MNG based on high
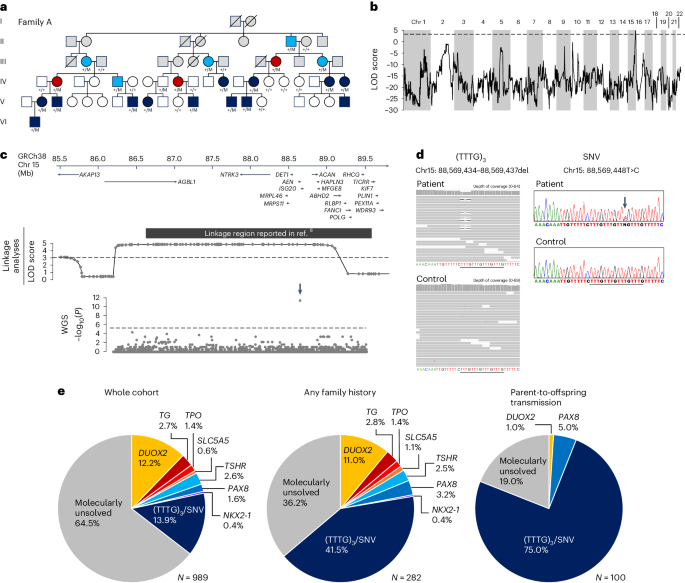
serum thyroglobulin levels (Fig. 1a). Linkage analysis identified a 3.2-Mb critical region on chr 15q26.1 (GRCh38 chr15: 86,206,051‒89,412,131, maximum logarithm of odds score = 4.8; Fig.
1b). The linkage region overlapped with a previously reported region linked to nongoitrous congenital hypothyroidism (Fig. 1c)8. The region encompasses 11 protein-coding genes, but no
candidate variant was found by exome sequencing (Supplementary Table 1). WGS OF UNSOLVED CONGENITAL HYPOTHYROIDISM FAMILIES We hypothesized that family A and a subset of the molecularly
unsolved congenital hypothyroidism families would have common or similar genetic change(s) in noncoding sequences within the linkage region. We selected ten unsolved congenital
hypothyroidism families (23 patients; Extended Data Fig. 2) for the next genetic investigation, which are as follows: three families with three or more affected members, one parent-offspring
pair and six sibling pairs. These ten families and family A were subject to WGS. As a control, 56 healthy Japanese individuals were sequenced. In the 3.2 Mb linkage region, we identified
2,446 rare variants, defined by allele frequency <0.002 in Tohoku Medical Megabank Organization (ToMMo)11, in the 81 participants. The linkage region was divided into 6,413 regions of 500
bp in size, and for each region, the density of rare variants was compared between the congenital hypothyroidism and control groups. We identified a single 500-bp region with a high density
of rare variants in the congenital hypothyroidism group (Fig. 1c). In the region, an identical heterozygous 4-bp deletion (GRCh38 chr15: 88,569,434–88,569,437del) was found in 8 of the 11
unsolved families. The variant affects a (TTTG)4 microsatellite, reducing the number of repeats from four to three times (designated as (TTTG)3; Fig. 1d). This microsatellite is located on
the polyA tail of an _Alu_ element, which is conserved in some primates but is not observed in nonprimate mammals (Extended Data Fig. 3). (TTTG)3 is an ultrarare variant observed in 3 of
38,722 healthy Japanese individuals registered in 38KJPN. The variant has been observed in non-Japanese populations, but at low frequencies in all of them (Supplementary Table 2). An
increase in the number of TTTG repeat, namely (TTTG)5, is also registered in TOPMed Freeze 10 (ref. 12) and ChinaMAP13 (Supplementary Table 2). SCREENING IN THE CONGENITAL HYPOTHYROIDISM
COHORT To identify congenital hypothyroidism patients with (TTTG)3 in the entire patient cohort, we conducted a PCR-based screen in 964 patients who were not subject to WGS. Further, 121
patients (73 families) with (TTTG)3 and two patients with a previously unreported single-nucleotide variant (SNV; GRCh38 chr15: 88,569,448T>C, designated as SNV) affecting the TTTG
microsatellite were identified (Fig. 1d and Supplementary Fig. 1). In the whole congenital hypothyroidism patient cohort (_n_ = 989), (TTTG)3 and SNV accounted for 13.9% of cases (Fig. 1e),
which was significantly higher than the general population (_P_ < 10−300). In the subgroup of patients with family history, the proportion of variant carriers increased to 41.5%, and it
reached 75.0% when limited to patients with parent-to-offspring transmission (Fig. 1e). Of the 83 variant-carrying families, parental genotyping was performed in 51. In 50 of 51 analyzed
families, transmission of the variant from either the father or the mother was confirmed. De novo acquisition of (TTTG)3 was observed in one family (family 67; Supplementary Fig. 1). To test
whether inherited (TTTG)3 was derived from a common ancestor, we performed haplotype analysis. We found four distinct haplotypes segregated with (TTTG)3 (Supplementary Fig. 2), indicating
that (TTTG)3 has been acquired on at least four chromosomes independently. Notably, it shows that (TTTG)3 is not one of the surrogate variants, but that (TTTG)3 itself is the disease-causing
variant. CHILDHOOD PHENOTYPES Clinical phenotypes of the variant-carrying patients were relatively uniform. Serum TSH levels were moderately elevated, but free T4 levels were usually within
the reference interval (that is, compensated hypothyroidism; Fig. 2a). Elevated serum thyroglobulin levels were seen in almost all patients, but it was relatively mild compared to the
thyroid peroxidase defect and the dual oxidase 2 defect. In most cases, the size of the thyroid was slightly small as compared with the age-matched mean (Fig. 2b). Levels of thyroidal iodine
uptake were variable, including approximately 15% having higher levels than the upper limit of the reference interval (Fig. 2c). The patients usually require levothyroxine replacement
permanently with doses of 1.5‒2.5 μg kg−1 d−1 in adulthood (Extended Data Fig. 4)14. No common extrathyroidal complications were observed in the 137 variant-carrying patients. Uncommon
complications included intellectual disability with epilepsy (_n_ = 2) or without epilepsy (_n_ = 2), renal hypoplasia (_n_ = 1), congenital hydronephrosis (_n_ = 1), Gitelman syndrome (_n_
= 1) and ventricular septal defect (_n_ = 1). The clinical phenotypes of the variant carriers were relatively uniform within the families, as exemplified by the serum TSH levels of
childhood-onset patients in family A (Extended Data Fig. 5). Two variant-carrying childhood-onset patients were untreated or discontinued treatment. The untreated patient was diagnosed in
the newborn period but was followed without treatment because free T4 levels were normal despite high serum TSH levels (14.0‒58.2 mU l−1; reference 0.5‒5.0). In this patient, palpation
revealed goiter at age 12 years and then it progressed. Levothyroxine replacement therapy was initiated at the age of 16 years, and thyroid enlargement was controlled. The other
treatment-discontinued case was found to have a high serum TSH level (TSH: 19.9 mU l−1) in the newborn period and was treated with levothyroxine until 3 years of age when the treatment was
discontinued. Serum TSH levels during the untreated period ranged from 10.7 to 26.1 mU l−1. At age 5 years, goiter was noted on palpation. Ultrasonographic findings were consistent with MNG,
with the largest nodule measuring 18 mm in diameter. Subsequently, the goiter worsened and the nodules increased in size. Levothyroxine replacement therapy was resumed at the age of 9
years, and then thyroid enlargement and nodule growth were controlled. Of 137 variant-carrying patients, 6 had an additional heterozygous variant in autosomal recessive congenital
hypothyroidism-associated genes, including _DUOX2_, _SLC26A4_ and _TSHR_ (Supplementary Table 3). Although there was no significant difference in serum TSH levels between the 6 patients and
the remaining 131 patients, the highest thyroglobulin level (640 ng ml−1) in infancy was observed in the patient who was heterozygous for (TTTG)3 and _DUOX2_ p.Leu1160del15. ADULT PHENOTYPES
Through family analysis, additional 76 variant carriers were found, including 58 individuals born before 1979 when newborn screening for congenital hypothyroidism was implemented in Japan.
Homozygous (TTTG)3 was found in one female patient who was diagnosed with hypothyroidism (TSH: 21.2 mU l−1, free T4: 0.89 ng dl−1) with MNG at the age of 58 years (family 32, the proband’s
maternal grandmother; Fig. 3a and Supplementary Figs. 1 and 3). She underwent fine needle aspiration cytology on the largest nodule (size, 13 × 12 × 16 mm), and the result was benign. Except
for the thyroid abnormalities, she had no relevant medical history. Of the 58 adult variant carriers born before 1979, 16 were receiving levothyroxine treatment. In 20 of the 42 untreated
individuals, thyroid function test showed high serum TSH in 35%, low serum free T4 in 0% and high serum thyroglobulin in 94% (Fig. 3a). We also analyzed serum samples, stored in the biobank,
derived from three (TTTG)3 carriers registered in ToMMo JPN38K, and found similar biochemical characteristics in them (Fig. 3a). Fourteen of 15 untreated adults who underwent thyroid
ultrasonography had MNG (Supplementary Fig. 3). A characteristic increase in blood flow was observed in most cases where blood flow was evaluated (Fig. 3b). Due to the large thyroid nodules,
three patients received thyroidectomy, and we could obtain one specimen (Supplementary Fig. 1; family 47, the proband’s maternal uncle; Fig. 3c (left)). Histological analysis revealed a
nodule clearly demarcated from the adjacent thyroid tissue by a well-formed sclerotic capsule (Fig. 3c (middle)). The follicles in the nodule were heterogeneous in size, and the number of
resorptive vacuoles was increased (Fig. 3c (right). No evidence of malignancy was seen. We tested whether (TTTG)3 and SNV were observed in an independent MNG cohort without a history of
childhood-onset hypothyroidism. By sequencing 33 patients with MNG followed at Kuma Hospital, Kobe, we identified two patients with (TTTG)3 and one with SNV (Supplementary Fig. 1). This
frequency (3 in 33) was significantly higher than the general Japanese population (_P_ < 1.2 × 10−8). FUNCTIONAL ANALYSES There are >600,000 microsatellites in the human genome16,17.
Variation in the number of microsatellite repeats is often observed, and most of them are considered to have negligible biological effects. We first questioned whether the region containing
the disease-associated microsatellite (designated as (TTTG)4) has epigenomic signatures as gene regulatory regions. Analysis of single-nucleus assay for transposase-accessible chromatin
(snATAC)–seq count data showed that the (TTTG)4-containing region is in an open chromatin configuration in the thyroid (Fig. 4a (yellow lines)). Comparison of snATAC–seq count data derived
from 154 cell types indicated that this configuration is highly selective for the thyroid (Fig. 4a (blue lines)). High-throughput chromosome conformation capture (Hi-C) experiment performed
in H1-hESC differentiated to definitive endoderm18 showed that the (TTTG)4 shares the same chromatin domain with three protein-coding genes (_MRPL46_, _MRPS11_ and _DET1_; Fig. 4a), all of
which are expressed ubiquitously (Extended Data Fig. 6). Altogether these experiments suggest that (TTTG)4-containing region could have the capacity to modulate gene expression, functioning
as a regulatory element. To this end, we performed a luciferase activity experiment including pGL4-(TTTG)4 reporters with or without the herpes simplex virus thymidine kinase promoter
(HSVTKp) and measured the luciferase activity in transiently transfected cells (rat thyroid cell line FRTL-5; Fig. 4b). Without the viral promoter, the pGL4-(TTTG)4 reporter showed activity
comparable to the empty vector. However, the reporter with the viral promoter (pGL4-(TTTG)4-HSVTKp) showed reduced HSVTKp-mediated expression of luciferase by 32 ± 6%, suggesting a repressor
activity (Fig. 4c). The variant reporters without HSVTKp (pGL4-(TTTG)3 and pGL4-SNV) showed substantial basal activities. However, these activities were not additive for HSVTKp; rather,
(TTTG)3 repressed the activity of HSVTKp. The degree of repression was attenuated as compared to that of the repression by (TTTG)4 (Fig. 4c). Similar results were confirmed in experiments
using human embryonic kidney 293 cells (Extended Data Fig. 7). Taken together, these bioinformatic and experimental data suggest that the (TTTG)4-containing region is a thyroid-specific
repressor, and sequence changes affecting the TTTG microsatellite cause loss of the repressor activity. DISCUSSION Here we showed that about three-quarters of cases of dominantly inherited
Japanese congenital hypothyroidism are caused by nucleotide changes affecting a noncoding microsatellite in 15q26.1. The untreated or treatment-discontinued cases showed a characteristic
transition of the clinical manifestation from nongoitrous congenital hypothyroidism to MNG, linking the two thyroid abnormalities that had been thought to be distinct. This genetic disease
has been registered in the Online Mendelian Inheritance in Man as congenital hypothyroidism, nongoitrous, 3 (CHNG3; %609893), also referred to as resistance to TSH8. However, we observed no
clinical evidence of defective TSH signaling in variant carriers, but rather overcompensation of the signal was presumed—goiter, increased blood flow and increased thyroidal iodine uptake.
In the future, more appropriate disease names would be assigned based on precise molecular pathogenesis. CHNG3 accounted for 13.9% (95% confidence interval, 11.9‒16.1) of our Japanese
congenital hypothyroidism cohort and is one of the leading genetic causes of congenital hypothyroidism in Japan (Fig. 1e). This study further showed that approximately 9% of Japanese MNG
were attributed to CHNG3, but given the small size of the patient cohort (_n_ = 33), the frequency should be validated in a larger cohort. Universal genetic screening for CHNG3 in patients
with MNG may be considered if significant clinical characteristics, such as risk of malignant transformation, are identified. Based on the ToMMo 38KJPN data and analysis of stored samples,
we estimated the prevalence of CHNG3 in Japan as 1/12,900 (95% confidence interval, 1/5,400 to 1/35,500). The absence of sequelae of congenital hypothyroidism such as intellectual disability
and short stature in untreated adult patients with CHNG3 is likely due to thyroid hormone production via increase of thyroid tissues. In other Mendelian forms of nongoitrous congenital
hypothyroidism, including _TSHR_ defect19, _PAX8_ defect20 and _NKX2-1_ defect21, goiter is not usually observed in untreated patients with high serum TSH levels (Extended Data Fig. 8).
Therefore, factor(s) other than compensatory TSH stimulation may be involved in the development of goiter and MNG in untreated patients with CHNG3. Although there are no obvious sequelae of
congenital hypothyroidism in untreated adult patients with CHNG3, levothyroxine therapy from infancy may have a role in preventing the transition from nongoitrous congenital hypothyroidism
to MNG and the resultant need for a fine needle aspiration biopsy and surgery. The adult untreated individual with homozygous (TTTG)3 had a higher serum TSH level than heterozygous variant
carriers and had a normal-sized thyroid gland despite the presence of multiple nodules. These observations suggest that the two (TTTG)3 alleles cause a more severe reduction in the
proliferation capacity of thyroid follicular cells than one allele but do not have catastrophic effects on thyroid development and physiology. In terms of methods to identify Mendelian
disease-causing nucleotide changes in noncoding regions, one or more genetic mapping methods such as analysis of structural variants22,23,24,25 and linkage analysis (this study and refs.
24,25) were used. Because exome sequencing has been popularized, most genetic studies no longer require these mapping methods to identify disease-causing variants in exons. However, given
the vastness of the noncoding regions (50 times larger than the coding regions), genetic mapping methods still have an indispensable role in identifying disease-causing noncoding variants.
The disease-causing noncoding variants identified in this study were located inside an _Alu_ element. Recent studies have shown that _Alu_ elements are involved in epigenomic modifications26
and 3D genome conformations27 in vitro. Genetic variants of _Alu_ associated with altered gene expression levels have been reported28, and there is growing interest in the impact of these
primate-specific repetitive sequences on the human genome. In our in vitro experiments, the variant luciferase reporters (pGL4-(TTTG)3 and pGL4-SNV) had enhancer-like activities, while
nonmutated reporter with the viral promoter (pGL4-(TTTG)4-HSVTKp) showed a repressor-like activity. In ref. 28, a comprehensive analysis of the effects of mobile elements, including _Alu_,
on gene expression regulation was performed to observe both positive and negative effects, with negative effects being more common. We assume that the _Alu_ element with (TTTG)4 was
originally acquired as a repressor during primate evolution to optimize thyroid gland anatomy and physiology, but its repressor activity was altered by the shortening of the TTTG repeat. Two
large meta-genome-wide association studies (GWAS) have reported single-nucleotide polymorphisms (SNPs) near (TTTG)4 associated with thyroid function—rs17776563 (distance, 6.5 kb)29 and
rs1348005 (distance, 0.8 kb)30 (Fig. 4a). According to the Genotype-Tissue Expression (GTEx) project V8 dataset, these two SNPs are expression quantitative trait loci that affect thyroidal
expression of _DET1_ (Supplementary Fig. 4), encoding a protein involved in proteasomal degradation of c-Jun31. Carriers of the low thyroid function-associated genotypes (rs17776563 A and
rs1348005 G) showed high thyroidal _DET1_ expression (Supplementary Fig. 4), suggesting that increased expression of _DET1_ and resultant decrease of c-Jun would negatively affect the
hormone-producing capacity of the thyroid. This assumption agrees well with the known role of the IGF1-Ras-MAPK/c-Jun signaling pathway in thyroid cell growth32,33. At present, we cannot
pinpoint which gene(s) are repressed by the (TTTG)4-containing region, but _DET1_ would be the most likely target. Further studies, such as RNA-seq of patient-derived thyroid tissues, are
needed to clarify the molecular mechanism(s) linking the noncoding changes and CHNG3. In summary, through linkage analysis of a large family and genetic screens in patient cohorts, we have
shown that nucleotide changes in a microsatellite located in the noncoding region of chr 15q26.1 are responsible for childhood nongoitrous congenital hypothyroidism and adult MNG, which have
been considered distinct disease entities. These findings provide unique insights into the relationship between the anatomy and physiology of the thyroid gland. METHODS ETHICAL
CONSIDERATION This study was approved by the Ethics Committees of Keio University School of Medicine (approval: 20140289 and 20170130), the National Center for Child Health and Development
(approval: 553) and ToMMo (approval: 2022-4-186). This study was conducted in accordance with the Declaration of Helsinki. All participants or their parents provided written informed consent
for the molecular studies. STUDY PARTICIPANTS AND GENETIC SCREENING In this study, congenital hypothyroidism was defined as patients with a positive newborn screening result and at least
one high serum TSH level by 2 months of age. Cases due to maternal Graves’ disease, exposure to antithyroid drug or exposure to excessive iodine were excluded. Family history was considered
positive if first- to third-degree relatives had hypothyroidism of any type. Patients were clinically characterized at each institution. Goitrous congenital hypothyroidism was defined as
cases with in situ thyroid gland of which size s.d. score34 was equal to or more than +2.0. From 2006 to 2022, 989 peripheral blood samples derived from congenital hypothyroidism patients
(female, 53%) were collected from 119 institutions across Japan. Eleven congenital hypothyroidism-associated genes (_DUOX2, DUOXA2, FOXE1, IYD, NKX2-1, PAX8, SLC5A5, SLC26A4, TG, TPO_ and
_TSHR_) were sequenced as previously described35,36. Detected variants were evaluated based on frequencies in the patient cohort (_n_ = 989) versus the general Japanese population (ToMMo
38KJPN, _n_ = 38,722), presumed functional impact and/or the results of cell-based experiments as previously described6,7,15. For the genetic diagnoses, the mode of inheritance and results
of parental genotyping, if available, were considered. Patients with MNG (_n_ = 33, female = 70%, median age = 48 years and interquartile range = 35–63 years) were clinically characterized
and enrolled at Kuma Hospital, Kobe city. The definition of MNG was based on the following ultrasonographic findings: (i) enlarged thyroid gland based on ultrasonographic measurements and
(ii) multiple thyroid nodules. THYROID ULTRASONOGRAPHY Thyroid ultrasonography was performed for 16 individuals with (TTTG)3 at each institution to evaluate the morphology of the thyroid
gland. Color-flow Doppler imaging, a noninvasive technique to visualize blood flow, was conducted in 14 of the 16 individuals. THYROID HISTOLOGY AND IMMUNOHISTOCHEMISTRY A thyroid tissue
sample was obtained from a (TTTG)3-carrying individual undergoing thyroidectomy (Supplementary Fig. 1; family 47, the proband’s maternal uncle). The sample was fixed in 10% formalin and
embedded in paraffin. Tissue sections were cut at 5 μm thickness, deparaffinized and stained with hematoxylin and eosin using standard procedures. For immunohistochemical staining of
thyroglobulin, 5-μm sections of the formalin-fixed paraffin-embedded thyroid tissue were deparaffinized, and endogenous peroxidase activity was blocked by incubation in 1% H2O2 to unmask
antigens. Then, the slides were microwaved in 10 mM citrate buffer (pH 6) for 15 min. After blocking nonspecific staining with horse serum, the slides were incubated with rabbit
antithyroglobulin polyclonal antibody (Dako) at 1:5,000 dilution. Sections were then incubated with biotin-labeled secondary antibody (dilution, 1:500). Subsequent reactions with the
ENVISION system (Dako) and diaminobenzidine (Sigma-Aldrich) were followed by counterstaining with hematoxylin. LINKAGE ANALYSIS SNPs of 13 individuals belonging to family A were genotyped
with GeneChip Human Mapping 250 K Nsp Array (Thermo Fisher Scientific). Linkage analysis was performed with Superlink Online SNP version 1.1 (ref. 37) to calculate multipoint logarithm of
the odds scores. We performed affected-only analysis (see Supplementary Fig. 2 for analyzed participants) with a 100% penetrant autosomal dominant model and an assumption of no phenocopies.
Disease allele frequency was set to 0.0001. WGS We performed WGS on 25 patients with unsolved familial congenital hypothyroidism. We also sequenced 56 ancestry-matched controls from the
National Center Biobank Network resource38 using the identical analytical pipeline. DNA libraries were constructed with TruSeq DNA PCR-Free (Illumina) and sequenced with NovaSeq6000
(Illumina) or DNB-SEQ (MGI). Read mapping to GRCh38 and variant calling were conducted with DRAGEN v3.9.5 (Illumina). In this study, rare variants were defined as allele frequency <0.002
in 38KJPN. The 3.2-Mb linkage region (GRCh38 chr15: 86,206,001–89,412,500) was divided into 6,413 regions with a size of 500 bp, and rare variants were counted for the patient and control
groups. Fisher’s exact test was used to compare the density of rare variants in each region with the Bonferroni-corrected significance threshold at _P_ = 7.8 × 10−6 (=0.05/6,413). PCR-BASED
SCREENING A 455-bp region (GRCh38 chr15: 88,569,231–88,569,685) was subject to PCR-based Sanger sequencing in patients with congenital hypothyroidism and MNG. Genetic and biochemical
analyses for family members of the variant-carrying patients were performed if consent for the study was obtained. The sequences of primers are shown in Supplementary Table 4. HAPLOTYPE
ANALYSIS (TTTG)3-carrying patients and their relatives were genotyped for five short-tandem repeat markers (D15S0299i, D15S199, D15S979, D15S0407i and D15S0496i) and four SNPs (rs201709422,
rs1348002, rs61650474 and rs191942900) with use of the fragment analysis method and PCR-based next-generation sequencing, respectively (Supplementary Table 5). BIOBANK SAMPLE ANALYSIS ToMMo
conducted WGS in 38,722 healthy Japanese individuals, and the summary statistics is publicly available as 38KJPN (https://jmorp.megabank.tohoku.ac.jp/). In the biobank cohort, (TTTG)3 was
observed in three individuals. Using the frozen serum samples derived from the three individuals, we measured levels of TSH (Lumipulse Presto TSH IFCC; Fujirebio), free T4 (Lumipulse Presto
FT4; Fujirebio), thyroglobulin (Lumipulse Presto iTACT Tg; Fujirebio), antithyroglobulin antibody (Lumipulse Presto TgAb; Fujirebio) and antithyroid peroxidase antibody (Lumipulse Presto
TPOAb; Fujirebio). HI-C We used Hi-C data of H1-hESC differentiated to definitive endoderm (accession: 4DNESCOQ5YRS; 4D Nucleome Data Portal) that was generated in the 4D nucleome project18.
Using the Hi-C data, 5-kb resolution contact matrices corresponding to chr15: 87,600,000–89,600,000 were visualized using Juicebox Web App (v2.3.5)39. SNATAC–SEQ snATAC–seq read count data
of 154 human cell types, generated in ref. 40, were downloaded from Human Enhancer Atlas (http://catlas.org/humanenhancer/). The obtained data (bigWig format) were analyzed with Megadepth
(version 1.2.0)41 to calculate regional mean read depth in 100-bp bins for each of the 154 cell types. Thyroid selectivity of snATAC–seq was defined by (median read depth of thyroid
follicular cell)/(sum of median read depth of 154 cell types). Read depth in the thyroid and thyroid selectivity were visualized with Microsoft Excel 2019. LUCIFERASE REPORTER ASSAY A 472-bp
genomic region (GRCh38 chr15: 88,569,213–88,569,684) containing the disease-associated microsatellite was PCR amplified. We cloned the PCR product into pGL4.23 (Promega), which contains the
minimal promoter and firefly luciferase sequences (pGL4-(TTTG)4) using the Gibson assembly technique (NEBuilder HiFi DNA Assembly Master Mix; New England Biolabs). Two disease-associated
variants were introduced with a standard site-directed mutagenesis technique (pGL4-(TTTG)3 and pGL4-SNV). These reporter vectors were used to evaluate the ability of the 472-bp sequence to
enhance gene expression. We also created vectors in which the minimal promoter was replaced by HSVTKp (pGL4-(TTTG)4-HSVTKp; Supplementary Methods), which were used to evaluate the ability of
the 472-bp sequence to repress gene expression. The complete sequences of the luciferase reporter vectors are shown in Supplementary Methods. Rat thyroid FRTL-5 cells were cultured in
Coon’s modified Ham’s F-12 medium supplemented with 5% bovine serum and a mixture of six hormones (10 μg ml−1 insulin, 0.36 ng ml−1 hydrocortisone, 5 μg ml−1 transferrin, 10 ng ml−1
somatostatin, 2 ng ml−1 glycyl-l-histidyl-l-lysine acetate and 1 mU ml−1 TSH) as previously described42. Cells seeded into six-well plates were transfected with 1 μg of each luciferase
reporter plasmid using FuGENE HD Transfection Reagent (Promega). Forty-eight hours after the transfection, luciferase assays were performed with cells lysed in Glo Lysis Buffer (Promega) and
Bright-Glo Luciferase Assay System (Promega). Protein concentrations of each cell lysate were measured with the DC Protein Assay Kit (Bio-Rad Laboratories), and the luminescence levels were
normalized. The luciferase activities are reported relative to the activity of the empty pGL4.23 vector (that is, fold activation). The data are representative of three independent
transfection experiments each performed as hexaplicate (FRTL-5) or octuplicate (HEK293). Two-sided Welch’s _t_ test was used for statistical comparisons. STATISTICS AND REPRODUCIBILITY This
is a cross-sectional observational study. No statistical method was used to predetermine sample size as this study involved a rare congenital disease. Histological studies were performed on
a resected thyroid sample from one individual. The study was conducted once due to limited sample availability. For the magnified view, representative images of four independent fields are
shown. Luciferase assays were performed three times as independent transfection experiments, and similar results were reproduced. No data were excluded from any of the experiments described.
Data collection and analysis were not performed blind to the conditions of the experiments. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio
Reporting Summary linked to this article. DATA AVAILABILITY Hi-C data of H1-hESC differentiated to definitive endoderm (accession 4DNESCOQ5YRS; 4D Nucleome Data Portal,
https://data.4dnucleome.org/experiment-set-replicates/4DNESCOQ5YRS/) were visualized using Juicebox Web App (https://aidenlab.org/juicebox/). snATAC–seq read count data were retrieved from
Human Enhancer Atlas (http://catlas.org/humanenhancer/)40. Frequency data of genetic variants in 38,722 healthy Japanese individuals (38KJPN) were obtained from jMorp
(https://jmorp.megabank.tohoku.ac.jp/). Expression quantitative trait locus data (GTEx v8 dataset) were downloaded from the GTEx portal (https://gtexportal.org/home/datasets). To preserve
the confidentiality of the study participants, restrictions apply to the use of the WGS data generated in this study. The corresponding author will, upon request, provide details of the
restrictions and the conditions under which access to some of the data may be provided. Source data are provided with this paper. CODE AVAILABILITY All software used in the study are
publicly available as described in Methods and the Reporting Summary. REFERENCES * Grosse, S. D. & Van Vliet, G. Prevention of intellectual disability through screening for congenital
hypothyroidism: how much and at what level? _Arch. Dis. Child._ 96, 374–379 (2011). Article PubMed Google Scholar * Dussault, J. H. et al. Preliminary report on a mass screening program
for neonatal hypothyroidism. _J. Pediatr._ 86, 670–674 (1975). Article CAS PubMed Google Scholar * Ieiri, T. et al. A 3′ splice site mutation in the thyroglobulin gene responsible for
congenital goiter with hypothyroidism. _J. Clin. Invest._ 88, 1901–1905 (1991). Article CAS PubMed PubMed Central Google Scholar * Abramowicz, M. J. et al. Identification of a mutation
in the coding sequence of the human thyroid peroxidase gene causing congenital goiter. _J. Clin. Invest._ 90, 1200–1204 (1992). Article CAS PubMed PubMed Central Google Scholar *
Moreno, J. C. et al. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. _N. Engl. J. Med._ 347, 95–102 (2002). Article CAS PubMed Google
Scholar * Narumi, S. et al. TSHR mutations as a cause of congenital hypothyroidism in Japan: a population-based genetic epidemiology study. _J. Clin. Endocrinol. Metab._ 94, 1317–1323
(2009). Article CAS PubMed Google Scholar * Narumi, S., Muroya, K., Asakura, Y., Adachi, M. & Hasegawa, T. Transcription factor mutations and congenital hypothyroidism: systematic
genetic screening of a population-based cohort of Japanese patients. _J. Clin. Endocrinol. Metab._ 95, 1981–1985 (2010). Article CAS PubMed Google Scholar * Grasberger, H. et al.
Identification of a locus for nongoitrous congenital hypothyroidism on chromosome 15q25.3-26.1. _Hum. Genet._ 118, 348–355 (2005). Article CAS PubMed Google Scholar * Krohn, K. et al.
Molecular pathogenesis of euthyroid and toxic multinodular goiter. _Endocr. Rev._ 26, 504–524 (2005). Article CAS PubMed Google Scholar * Zu, T., Pattamatta, A. & Ranum, L. P. W.
Repeat-associated non-ATG translation in neurological diseases. _Cold Spring Harb. Perspect. Biol._ 10, a033019 (2018). Article CAS PubMed PubMed Central Google Scholar * Tadaka, S. et
al. jMorp updates in 2020: large enhancement of multi-omics data resources on the general Japanese population. _Nucleic Acids Res._ 49, D536–D544 (2021). Article CAS PubMed Google Scholar
* Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. _Nature_ 590, 290–299 (2021). Article CAS PubMed PubMed Central Google Scholar * Cao, Y. et al.
The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals. _Cell Res._ 30, 717–731 (2020). Article PubMed PubMed Central Google Scholar * Delvecchio, M. et al.
Levothyroxine requirement in congenital hypothyroidism: a 12-year longitudinal study. _Endocrine_ 50, 674–680 (2015). Article CAS PubMed Google Scholar * Narumi, S., Muroya, K., Asakura,
Y., Aachi, M. & Hasegawa, T. Molecular basis of thyroid dyshormonogenesis: genetic screening in population-based Japanese patients. _J. Clin. Endocrinol. Metab._ 96, E1838–E1842 (2011).
Article CAS PubMed Google Scholar * Borstnik, B. & Pumpernik, D. Tandem repeats in protein coding regions of primate genes. _Genome Res._ 12, 909–915 (2002). Article CAS PubMed
PubMed Central Google Scholar * Li, Y. C., Korol, A. B., Fahima, T. & Nevo, E. Microsatellites within genes: structure, function, and evolution. _Mol. Biol. Evol._ 21, 991–1007 (2004).
Article CAS PubMed Google Scholar * Dekker, J. et al. The 4D nucleome project. _Nature_ 549, 219–226 (2017). Article CAS PubMed PubMed Central Google Scholar * Alberti, L. et al.
Germline mutations of TSH receptor gene as cause of nonautoimmune subclinical hypothyroidism. _J. Clin. Endocrinol. Metab._ 87, 2549–2555 (2002). Article CAS PubMed Google Scholar * De
Sanctis, L. et al. Familial PAX8 small deletion (c.989_992delACCC) associated with extreme phenotype variability. _J. Clin. Endocrinol. Metab._ 89, 5669–5674 (2004). Article PubMed Google
Scholar * Doyle, D. A., Gonzalez, I., Thomas, B. & Scavina, M. Autosomal dominant transmission of congenital hypothyroidism, neonatal respiratory distress, and ataxia caused by a
mutation of NKX2-1. _J. Pediatr._ 145, 190–193 (2004). Article CAS PubMed Google Scholar * Lettice, L. A. et al. A long-range Shh enhancer regulates expression in the developing limb and
fin and is associated with preaxial polydactyly. _Hum. Mol. Genet._ 12, 1725–1735 (2003). Article CAS PubMed Google Scholar * Wakeling, M. N. et al. Non-coding variants disrupting a
tissue-specific regulatory element in HK1 cause congenital hyperinsulinism. _Nat. Genet._ 54, 1615–1620 (2022). Article CAS PubMed PubMed Central Google Scholar * Tenney, A. P. et al.
Noncoding variants alter GATA2 expression in rhombomere 4 motor neurons and cause dominant hereditary congenital facial paresis. _Nat. Genet._ 55, 1149–1163 (2023). Article CAS PubMed
PubMed Central Google Scholar * Small, K. W. et al. North Carolina Macular Dystrophy is caused by dysregulation of the retinal transcription factor PRDM13. _Ophthalmology_ 123, 9–18
(2016). Article PubMed Google Scholar * Ferrari, R. et al. TFIIIC binding to Alu elements controls gene expression via chromatin looping and histone acetylation. _Mol. Cell_ 77, 475–487
(2020). Article CAS PubMed PubMed Central Google Scholar * Liang, L. et al. Complementary Alu sequences mediate enhancer-promoter selectivity. _Nature_ 619, 868–875 (2023). Article CAS
PubMed Google Scholar * Kojima, S. et al. Mobile element variation contributes to population-specific genome diversification, gene regulation and disease risk. _Nat. Genet._ 55, 939–951
(2023). Article CAS PubMed Google Scholar * Porcu, E. et al. A meta-analysis of thyroid-related traits reveals novel loci and gender-specific differences in the regulation of thyroid
function. _PLoS Genet._ 9, e1003266 (2013). Article CAS PubMed PubMed Central Google Scholar * Zhou, W. et al. GWAS of thyroid stimulating hormone highlights pleiotropic effects and
inverse association with thyroid cancer. _Nat. Commun._ 11, 3981 (2020). Article CAS PubMed PubMed Central Google Scholar * Wertz, I. E. et al. Human De-etiolated-1 regulates c-Jun by
assembling a CUL4A ubiquitin ligase. _Science_ 303, 1371–1374 (2004). Article CAS PubMed Google Scholar * Krieger, C. C., Morgan, S. J., Neumann, S. & Gershengorn, M. C. Thyroid
stimulating hormone (TSH)/insulin-like growth factor 1 (IGF1) receptor cross-talk in human cells. _Curr. Opin. Endocr. Metab. Res._ 2, 29–33 (2018). Article PubMed PubMed Central Google
Scholar * Muller, K. et al. TSH compensates thyroid-specific IGF-I receptor knockout and causes papillary thyroid hyperplasia. _Mol. Endocrinol._ 25, 1867–1879 (2011). Article PubMed
PubMed Central Google Scholar * Ejiri, H. et al. Ultrasonography-based reference values for the cross-sectional area of the thyroid gland in children and adolescents: the Fukushima Health
Management Survey. _Clin. Pediatr. Endocrinol._ 32, 52–57 (2023). Article PubMed Google Scholar * Sugisawa, C. et al. Adult thyroid outcomes of congenital hypothyroidism. _Thyroid_ 33,
556–565 (2023). Article CAS PubMed Google Scholar * Yoshizawa-Ogasawara, A., Ogikubo, S., Satoh, M., Narumi, S. & Hasegawa, T. Congenital hypothyroidism caused by a novel mutation of
the dual oxidase 2 (_DUOX2_) gene. _J. Pediatr. Endocrinol. Metab._ 26, 45–52 (2013). Article CAS PubMed Google Scholar * Silberstein, M. et al. A system for exact and approximate
genetic linkage analysis of SNP data in large pedigrees. _Bioinformatics_ 29, 197–205 (2013). Article CAS PubMed Google Scholar * Kawai, Y. et al. Exploring the genetic diversity of the
Japanese population: insights from a large-scale whole genome sequencing analysis. _PLoS Genet._ 19, e1010625 (2023). Article CAS PubMed PubMed Central Google Scholar * Durand, N. C. et
al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. _Cell Syst._ 3, 99–101 (2016). Article CAS PubMed PubMed Central Google Scholar * Zhang, K. et
al. A single-cell atlas of chromatin accessibility in the human genome. _Cell_ 184, 5985–6001 (2021). Article CAS PubMed PubMed Central Google Scholar * Wilks, C. et al. Megadepth:
efficient coverage quantification for BigWigs and BAMs. _Bioinformatics_ 37, 3014–3016 (2021). Article CAS PubMed PubMed Central Google Scholar * Suzuki, K. et al. Autoregulation of
thyroid-specific gene transcription by thyroglobulin. _Proc. Natl Acad. Sci. USA_ 95, 8251–8256 (1998). Article CAS PubMed PubMed Central Google Scholar Download references
ACKNOWLEDGEMENTS We thank all the patients and their families for participating in the study. We thank the following primary physicians who clinically evaluated patients: M. Adachi and Y.
Asakura (Department of Endocrinology and Metabolism, Kanagawa Children’s Medical Center); K. Aizu (Division of Endocrinology and Metabolism, Saitama Children’s Medical Center); D. Ariyasu
(Department of Pediatrics, Kawasaki Municipal Hospital); T. Hamajima and M. Kimura (Department of Endocrinology and Metabolism, Aichi Children’s Health and Medical Center); T. Hotsubo
(Department of Pediatrics, Sapporo Children’s Endocrine Clinic); S. Ida, T. Wada and M. Kawai (Department of Gastroenterology and Endocrinology, Osaka Women’s and Children’s Hospital); M.
Honda, Y. Ichihashi, M. Inokuchi and T. Sato (Department of Pediatrics, Keio University School of Medicine); A. Ishii and H. Kamasaki (Department of Pediatrics, Sapporo Medical University);
T. Kasahara and S. Fukata (Department of Internal Medicine, Kuma Hospital); K. Kashimada (Department of Pediatrics and Developmental Biology, Tokyo Medical and Dental University); K. Kitsuda
(Department of Pediatrics, Kitasato University School of Medicine); H. Kobayashi (Central Clinical Laboratory, Shimane University Hospital); Y. Komori (Department of Pediatrics, Wakayama
Rosai Hospital); T. Mochizuki (Kibounomori Growth and Development Clinic); Y. Baba, M. Mitani-Konno, A. Shimada and H. Zukeran (Division of Endocrinology and Metabolism, Tokyo Metropolitan
Children’s Medical Center); Y. Mushimoto (Department of Endocrinology and Metabolism, Fukuoka Children’s Hospital); Y. Naiki (Department of Endocrinology and Metabolism, National Center for
Child Health and Development); J. Nishioka (Department of Pediatrics, Public Yame General Hospital); C. Numakura and A. Muranaka (Department of Pediatrics, Yamagata University School of
Medicine); S. Okada (Department of Pediatrics, Hiroshima University Graduate School of Biomedical and Health Sciences); M. Okajima (Department of Pediatrics, School of Medicine, Institute of
Medical, Pharmaceutical and Health Sciences, Kanazawa University); T. Saito (Department of Pediatrics, Matsudo City General Hospital); K. Sawano (Division of Pediatrics, Department of
Homeostatic Regulation and Development, Niigata University Graduate School of Medical and Dental Sciences); H. Shinohara (Department of Pediatrics, Ibaraki Seinan Medical Center Hospital);
S. Soneda (Department of Pediatrics, St. Marianna University School of Medicine); T. Tajima (Department of Pediatrics, Jichi Medical University); H. Tanaka and S. Kitanaka (Department of
Pediatrics, University of Tokyo); N. Uchida (Department of Pediatrics, Saiseikai Utsunomiya Hospital); T. Urakami and Y. Mine (Department of Pediatrics, Nihon University School of Medicine).
We also thank H. Ogata-Kawata (Department of Maternal-Fetal Biology, National Research Institute for Child Health and Development), S. Miyasako, I. Kageyama and E. Suzuki (Department of
Molecular Endocrinology, National Research Institute for Child Health and Development) for their technical assistance; R. Katoh (Department of Pathology, Ito Hospital) and M. Hirokawa
(Department of Diagnostic Pathology and Cytology, Kuma Hospital) for preparation and evaluation of pathological samples; K. Suzuki (Department of Clinical Laboratory Science, Faculty of
Medical Technology, Teikyo University), T. Mizuguchi and N. Matsumoto (Department of Human Genetics, Yokohama City University Graduate School of Medicine) for fruitful discussion. We also
thank all the volunteers who participated in the Tohoku Medical Megabank (TMM) Project and the TMM Project Study Group. This work was supported by JSPS KAKENHI (grants JP23H02885 (to S.N.),
JP23K15400 (to M.K.) and JP21K07325 (to T.K.)), Japan Agency for Medical Research and Development (AMED; grants JP21tm0124005 (to J.T. and G.T.) and JP21tm0424601 (to J.T. and G.T.)) and
Takeda Science Foundation (to M.F., S.N.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * Department of Pediatrics, Keio University School of Medicine, Tokyo, Japan Satoshi Narumi, Kazuhiro Shimura, Kiyomi Abe, Chiho Sugisawa, Tomohiro Ishii &
Tomonobu Hasegawa * Department of Molecular Endocrinology, National Research Institute for Child Health and Development, Tokyo, Japan Satoshi Narumi, Erika Uehara, Kazuhisa Akiba, Kanako
Tanase-Nakao & Maki Fukami * Division of Pediatrics, Department of Homeostatic Regulation and Development, Niigata University Graduate School of Medical and Dental Sciences, Niigata,
Japan Keisuke Nagasaki * Department of Clinical Laboratory Science, Faculty of Medical Technology, Teikyo University, Tokyo, Japan Mitsuo Kiriya * Division of Endocrinology and Metabolism,
Tokyo Metropolitan Children’s Medical Center, Tokyo, Japan Kazuhisa Akiba & Yukihiro Hasegawa * Department of Internal Medicine, Ito Hospital, Tokyo, Japan Chiho Sugisawa & Natsuko
Watanabe * Department of Endocrinology and Metabolism, Fukuoka Children’s Hospital, Fukuoka, Japan Kenichi Miyako * Department of Pediatrics, Shiga University of Medical Science, Otsu, Japan
Yoshihiro Maruo * Department of Endocrinology and Metabolism, Kanagawa Children’s Medical Center, Yokohama, Japan Koji Muroya * Center for Excellence in Thyroid Care, Kuma Hospital, Kobe,
Japan Eijun Nishihara * Department of Genetic Diagnosis and Laboratory Medicine, Dokkyo Medical University, Mibu, Japan Yuka Ito & Takahiko Kogai * Department of Pathology, Showa
University Northern Yokohama Hospital, Yokohama, Japan Kaori Kameyama * Department of Maternal-Fetal Biology, National Research Institute for Child Health and Development, Tokyo, Japan
Kazuhiko Nakabayashi & Kenichiro Hata * Department of Human Molecular Genetics, Gunma University Graduate School of Medicine, Maebashi, Japan Kenichiro Hata * Department of Pediatrics,
Tohoku University Graduate School of Medicine, Sendai, Japan Hirohito Shima & Atsuo Kikuchi * Department of AI and Innovative Medicine, Tohoku University Graduate School of Medicine,
Sendai, Japan Jun Takayama & Gen Tamiya * Department of Integrative Genomics, Tohoku Medical Megabank Organization (ToMMo) Tohoku University, Sendai, Japan Jun Takayama & Gen Tamiya
* Statistical Genetics Team, RIKEN Center for Advanced Intelligence Project, Tokyo, Japan Jun Takayama & Gen Tamiya Authors * Satoshi Narumi View author publications You can also search
for this author inPubMed Google Scholar * Keisuke Nagasaki View author publications You can also search for this author inPubMed Google Scholar * Mitsuo Kiriya View author publications You
can also search for this author inPubMed Google Scholar * Erika Uehara View author publications You can also search for this author inPubMed Google Scholar * Kazuhisa Akiba View author
publications You can also search for this author inPubMed Google Scholar * Kanako Tanase-Nakao View author publications You can also search for this author inPubMed Google Scholar * Kazuhiro
Shimura View author publications You can also search for this author inPubMed Google Scholar * Kiyomi Abe View author publications You can also search for this author inPubMed Google
Scholar * Chiho Sugisawa View author publications You can also search for this author inPubMed Google Scholar * Tomohiro Ishii View author publications You can also search for this author
inPubMed Google Scholar * Kenichi Miyako View author publications You can also search for this author inPubMed Google Scholar * Yukihiro Hasegawa View author publications You can also search
for this author inPubMed Google Scholar * Yoshihiro Maruo View author publications You can also search for this author inPubMed Google Scholar * Koji Muroya View author publications You can
also search for this author inPubMed Google Scholar * Natsuko Watanabe View author publications You can also search for this author inPubMed Google Scholar * Eijun Nishihara View author
publications You can also search for this author inPubMed Google Scholar * Yuka Ito View author publications You can also search for this author inPubMed Google Scholar * Takahiko Kogai View
author publications You can also search for this author inPubMed Google Scholar * Kaori Kameyama View author publications You can also search for this author inPubMed Google Scholar *
Kazuhiko Nakabayashi View author publications You can also search for this author inPubMed Google Scholar * Kenichiro Hata View author publications You can also search for this author
inPubMed Google Scholar * Maki Fukami View author publications You can also search for this author inPubMed Google Scholar * Hirohito Shima View author publications You can also search for
this author inPubMed Google Scholar * Atsuo Kikuchi View author publications You can also search for this author inPubMed Google Scholar * Jun Takayama View author publications You can also
search for this author inPubMed Google Scholar * Gen Tamiya View author publications You can also search for this author inPubMed Google Scholar * Tomonobu Hasegawa View author publications
You can also search for this author inPubMed Google Scholar CONTRIBUTIONS S.N. and T.H. conceptualized the experimental design. K. Nagasaki and S.N. performed linkage analysis. S.N. and
K.T.-N. performed haplotype analysis. E.U., M.K. and K. Akiba performed in vitro experiments. S.N., E.U., K.T.-N., K.S., K. Abe, C.S., E.N., Y.I. and T.K. conducted genetic analysis. T.I.,
K. Miyako, Y.H., Y.M., K. Muroya, N.W. and E.N. clinically characterized patients and collected biological samples. M.F. coordinated the storage of biological samples. K.K. performed
pathological analysis. S.N., K. Nakabayashi and K.H. performed WGS. H.S., A.K., J.T. and G.T. analyzed biobank samples. S.N. supervised the overall study and wrote the initial paper. All
authors read, had the opportunity to comment on and approved the final paper. CORRESPONDING AUTHOR Correspondence to Satoshi Narumi. ETHICS DECLARATIONS COMPETING INTERESTS The authors
declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Genetics_ thanks Andrea Cortese, Luca Persani, Val Sheffield, and the other, anonymous, reviewer(s) for their
contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. EXTENDED DATA EXTENDED DATA FIG. 1 MOLECULAR DIAGNOSIS OF JAPANESE CONGENITAL HYPOTHYROIDISM. Results of targeted next-generation sequencing of known causative genes of
congenital hypothyroidism are shown. Even when restricted to patients with family history, about three-quarters of patients were not molecularly resolved. Source data EXTENDED DATA FIG. 2
PATIENTS ANALYZED BY LINKAGE ANALYSIS AND WHOLE-GENOME SEQUENCING (WGS). Pedigrees of 11 unsolved congenital hypothyroidism families subject to whole-genome sequencing are shown. Black
symbols indicate patients with congenital hypothyroidism, subclinical hypothyroidism or multinodular goiter (MNG). Daggers and asterisks denote individuals subject to linkage analysis and
WGS, respectively. EXTENDED DATA FIG. 3 GENOMIC LOCATION OF THE DISEASE-ASSOCIATED MICROSATELLITE. A, UCSC genome browser images showing the location of disease-associated microsatellite
(TTTG)4, sequence conservation in 27 primates and three non-primate mammals and repetitive sequences. Note that the microsatellite is part of an Alu element conserved in a subset of
primates. B, Comparison of the _AluSx1_ consensus sequence with the genomic sequence containing disease-associated microsatellite; the location of (TTTG)4, which corresponds to the polyA
tail of _AluSx1_, is highlighted in yellow. EXTENDED DATA FIG. 4 CHRONOLOGICAL CHANGES OF DAILY LEVOTHYROXINE DOSES IN COMPENSATED HYPOTHYROIDISM PATIENTS WITH (TTTG)3. The shaded area
indicates the range (−1 to +1 standard deviation) of ‘complete’ replacement obtained from congenital hypothyroidism patients with thyroid aplasia (ref. 11). Source data EXTENDED DATA FIG. 5
INTRAFAMILIAL VARIABILITY OF CLINICAL PHENOTYPES OF CONGENITAL HYPOTHYROIDISM PATIENTS WITH (TTTG)3. Left panel shows the pedigree of family A. Dark blue, light blue, red, white and gray
symbols indicate individuals with childhood-onset compensated hypothyroidism, subclinical hypothyroidism diagnosed in adulthood, suspected multinodular goiter (MNG), normal thyroid function
and unknown thyroid function, respectively. The numbers shown below the symbols indicate serum thyroid-stimulating hormone (TSH) levels evaluated after infancy with transient discontinuation
of levothyroxine treatment. NA denotes not available. Right panel shows the relatively uniform distribution of the serum TSH levels of the congenital hypothyroidism patients in family A.
EXTENDED DATA FIG. 6 RNA EXPRESSION LEVELS OF _MRPL46_, _MRPS11_ AND _DET1_ BY ORGANS. RNA expression values are shown in TPM (transcripts per million). Box plots are shown as median and
25th and 75th percentiles; points are displayed as outliers if they are above or below 1.5 times the interquartile range. Data were obtained from GTEx (https://gtexportal.org/). Each RNA
expression data was obtained from 4 to 803 donors (median 246). Arrows indicate thyroid. The three genes show ubiquitous gene expression. EXTENDED DATA FIG. 7 RESULTS OF LUCIFERASE ASSAYS
USING HEK293 CELLS. The 472 bp sequence exhibited an ability to repress herpes simplex virus thymidine kinase promoter (HSVTKp)-mediated transcription, while this repression was partially
lost by introduction of (TTTG)3 or the single nucleotide variant (SNV). Two-sided Welch’s t-test was used for statistical comparisons. Source data EXTENDED DATA FIG. 8 SCHEMATIC DIAGRAM
SHOWING AGE-DEPENDENT CHANGES IN THE CLINICAL PHENOTYPE OF CHNG3 (OMIM %609893). Patients with CHNG3 diagnosed through newborn screening, have mild thyroid hypoplasia and do not develop
thyroid nodules if treated with levothyroxine (LT4). If thyroid abnormalities are not diagnosed in infancy, thyroid hormone production is compensated via thyroid cell proliferation (that is,
goiter), eventually leading to multinodular goiter (MNG). LT4 treatment can prevent progression of goiter and further formation of nodules. MNG is also observed in childhood-onset patients
who stop treatment. This age-dependent transition of phenotypes has not been documented in other Mendelian forms of nongoitrous congenital hypothyroidism (CH), including TSHR defect, PAX8
defect and NKX2-1 defect, and thus is considered a clinical characteristic of CHNG3. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Methods and Figs. 1–4. REPORTING
SUMMARY SUPPLEMENTARY TABLES Supplementary Tables 1–5. SOURCE DATA SOURCE DATA FIG. 1 Statistical source data. SOURCE DATA FIG. 2 Statistical source data. SOURCE DATA FIG. 3 Statistical
source data. SOURCE DATA FIG. 3 Unprocessed images of ultrasonography and histology. SOURCE DATA FIG. 4 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 1 Statistical source data.
SOURCE DATA EXTENDED DATA FIG. 4 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 7 Statistical source data. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a
Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are
included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Narumi, S., Nagasaki, K., Kiriya, M. _et al._ Functional variants in
a TTTG microsatellite on 15q26.1 cause familial nonautoimmune thyroid abnormalities. _Nat Genet_ 56, 869–876 (2024). https://doi.org/10.1038/s41588-024-01735-5 Download citation * Received:
30 July 2023 * Accepted: 25 March 2024 * Published: 07 May 2024 * Issue Date: May 2024 * DOI: https://doi.org/10.1038/s41588-024-01735-5 SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative
Trending News
Aarp rewards: get ahead of whatever life has aheadMemorial Day Sale! Join AARP for just $11 per year with a 5-year membership Join now and get a FREE gift. Expires 6/4 G...
Equity indices subdued, banking stocks dip | dynamite newsEQUITY BENCHMARK INDICES WERE SUBDUED DURING EARLY HOURS ON TUESDAY DUE TO DRAG IN BANKING STOCKS AFTER THE RESERVE BANK...
Why aren't stroke patients getting clot-busting tpa drug?Memorial Day Sale! Join AARP for just $11 per year with a 5-year membership Join now and get a FREE gift. Expires 6/4 G...
IIS 10.0 Detailed Error - 404.0 - Not FoundHTTP Error 404.0 - Not Found The resource you are looking for has been removed, had its name changed, or is temporarily ...
Video 5: age-friendly network trainingMemorial Day Sale! Join AARP for just $11 per year with a 5-year membership Join now and get a FREE gift. Expires 6/4 G...
Latests News
Functional variants in a tttg microsatellite on 15q26. 1 cause familial nonautoimmune thyroid abnormalitiesABSTRACT Insufficient thyroid hormone production in newborns is referred to as congenital hypothyroidism. Multinodular g...
How One Woman Reinvented Her Career After 50AARP Foundation helped more than 1 million older adults living with low income secure more than $1 billion in income, be...
Rosalind Beck | TheArticleFirst {{register.errors.names}} Last Gender What's this for? Age bracket What's this for? This is to help us s...
Alzheimer’s prediction via smell and eye testsIn the search for easy-to-perform tests to predict Alzheimer’s disease risk, new research suggests the nose might know a...
Presence-absence of marine macrozoobenthos does not generally predict abundance and biomassABSTRACT Many monitoring programmes of species abundance and biomass increasingly face financial pressures. Occupancy is...