Exploring the genetic and genomic connection underlying neurodegeneration with brain iron accumulation and the risk for parkinson’s disease
Exploring the genetic and genomic connection underlying neurodegeneration with brain iron accumulation and the risk for parkinson’s disease"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Neurodegeneration with brain iron accumulation (NBIA) represents a group of neurodegenerative disorders characterized by abnormal iron accumulation in the brain. In Parkinson’s
Disease (PD), iron accumulation is a cardinal feature of degenerating regions in the brain and seems to be a key player in mechanisms that precipitate cell death. The aim of this study was
to explore the genetic and genomic connection between NBIA and PD. We screened for known and rare pathogenic mutations in autosomal dominant and recessive genes linked to NBIA in a total of
4481 PD cases and 10,253 controls from the Accelerating Medicines Partnership Parkinsons’ Disease Program and the UKBiobank. We examined whether a genetic burden of NBIA variants contributes
to PD risk through single-gene, gene-set, and single-variant association analyses. In addition, we assessed publicly available expression quantitative trait loci (eQTL) data through
Summary-based Mendelian Randomization and conducted transcriptomic analyses in blood of 1886 PD cases and 1285 controls. Out of 29 previously reported NBIA screened coding variants, four
were associated with PD risk at a nominal _p_ value < 0.05. No enrichment of heterozygous variants in NBIA-related genes risk was identified in PD cases versus controls. Burden analyses
did not reveal a cumulative effect of rare NBIA genetic variation on PD risk. Transcriptomic analyses suggested that _DCAF17_ is differentially expressed in blood from PD cases and controls.
Due to low mutation occurrence in the datasets and lack of replication, our analyses suggest that NBIA and PD may be separate molecular entities. SIMILAR CONTENT BEING VIEWED BY OTHERS
EXPANDING CAUSAL GENES FOR PARKINSON’S DISEASE VIA MULTI-OMICS ANALYSIS Article Open access 21 October 2023 GENETIC ANALYSES IDENTIFY CIRCULATING GENES RELATED TO BRAIN STRUCTURES ASSOCIATED
WITH PARKINSON’S DISEASE Article Open access 14 January 2025 LOSS-OF-FUNCTION VARIANTS IN _ITSN1_ CONFER HIGH RISK OF PARKINSON’S DISEASE Article Open access 15 August 2024 INTRODUCTION
Neurodegeneration with brain iron accumulation (NBIA) represents a group of inherited heterogeneous neurodegenerative disorders characterized by iron accumulation and the presence of axonal
spheroids in the basal ganglia and other brain areas1. The worldwide prevalence of NBIA in the general population is estimated between 1–3/1,000,000 individuals, adding these disorders to
the group of ultra-rare orphan diseases2. In view of the rarity of these disorders and its diverse clinical presentation, NBIA can often go unrecognized, underdiagnosed, or misdiagnosed. In
the brain, the substantia nigra, putamen, globus pallidus and caudate nucleus have the highest iron concentration, and total iron content increases with age3. Brain iron is crucial for
important processes such as the synthesis of myelin and neurotransmitters and oxygen transport3,4. Higher nigral iron concentrations have been shown in Parkinson’s Disease (PD) patients in
postmortem studies repeatedly5,6. This excess of intracellular iron is associated with oxidative stress, lipid peroxidation, cellular dysfunction, and neuronal death in PD and thus
potentially playing a role in PD pathogenesis7,8. To date, mutations in 13 genes have been associated with autosomal dominant (_FTL_), autosomal recessive (_ATP13A2, PANK2, PLA2G6, FA2H, CP,
C19orf12, COASY, GTPBP2, DCAF17, VAC14_) and X-linked forms of NBIA (_WDR45, RAB39B_), and are involved in a wide range of molecular processes affecting mitochondrial function, coenzyme A
metabolism, lipid metabolism and autophagy9. Typically, NBIA disorders present with variable and complex phenotypes including parkinsonism, dystonia, intellectual disability, and cognitive
decline1. The NBIA clinical spectrum is broad and there is increased awareness of clinical overlap between different NBIA disorders as well as with other diseases such as Parkinson’s disease
(PD). In PD, it has been widely suggested that genetic components contributing to disease might include rare genetic variants of small or moderate effect, where functional and deleterious
alleles might exist10. Furthermore, it has been previously predicted that variability in genes causing young-onset autosomal recessive neurological diseases contribute to late-onset
neurological diseases. In fact, homozygous and compound heterozygous loss-of-function mutations in _TREM2_ have been previously associated with an autosomal recessive form of early-onset
dementia, while when found in heterozygous state confer moderate risk for Alzheimer’s disease11,12. Similarly, homozygous and compound heterozygous mutations in _GBA_ are responsible for
Gaucher’s disease while heterozygous variants are a well-validated risk factor for PD13. In PD, iron accumulation is a cardinal feature of degenerating regions in the brain and has been
implicated in mechanisms that precipitate cell death14. Iron is a transition metal involved in several cellular functions such as neuronal metabolism, DNA synthesis, oxygen transport,
mitochondrial respiration, and myelination in the brain15. Furthermore, it is implicated in production and turnover of some neurotransmitters such as dopamine, epinephrine or serotonin16.
Iron tends to accumulate with age mainly in the cortex and the nuclei of the basal ganglia, including the globus pallidus, putamen, and caudate nucleus; and an excessive accumulation in
these regions is associated with neurodegenerative disorders17. Evidence suggests abnormal iron levels in the brains of PD patients and a role for iron dysregulation in the disease18.
Interestingly, NBIA-related genes are involved in mitochondrial function and autophagy, dysfunction of which is implicated in PD, suggesting there could be shared molecular pathways and gene
networks between NBIA and PD19. Furthermore, autopsy examination of NBIA-genetically confirmed cases has demonstrated Lewy bodies in some subforms, linking the pathology of both diseases20.
The aim of the present study was to explore the relationship between genes known to contain mutations that cause autosomal dominant/recessive NBIA and PD etiology. Using the largest genetic
and genomic datasets of PD cases and controls to date including the Accelerating Medicines Partnership Program for Parkinson’s Disease (AMP-PD) and the UKBiobank (UKB), we screened for the
presence of known and rare pathogenic mutations linked to NBIA in PD. We further examined whether a genetic burden of variants in NBIA-linked genes could contribute to the risk of developing
PD by performing single gene, gene-set, and single variant association analyses. Finally, to investigate the potential effect of changes in NBIA-linked genes expression in PD compared to
healthy individuals, we assessed publicly available expression quantitative trait loci (eQTL) results from GTEx v8, BRAINEAC, and transcriptomics data from AMP-PD. RESULTS GENETIC SCREENING
OF NBIA-RELATED GENES IN WHOLE-GENOME AND WHOLE-EXOME SEQUENCING DATA OF PARKINSON’S DISEASE CASES AND CONTROLS ATP13A2 Genetic variants in the _ATPase Cation Transporting 13A2_ (_ATP13A2_)
gene, located on chromosome 1, have been previously associated with Kufor-Rakeb syndrome, spastic paraplegia type 78, and parkinsonism21,22,23. A total of 36 protein-coding variants were
present in the screened datasets, of which four missense variants had a higher frequency in cases versus controls. A summary of variants with a higher frequency in cases can be found in
Table 1 while additional variant information is listed in the supplemental. The variants found in AMP-PD (p.I946F, p.A249V, p.T12M) have been previously associated with PD in individuals of
European ancestry although their pathogenicity is still unclear24,25. The p.T402M variant, found in UKB, has been previously described in a homozygous individual with ataxia-myoclonus
syndrome with an age at onset of 38 and lacking parkinsonism26,27. The report postulates that together, progressive ataxia with action myoclonus along with lack of parkinsonism may suggest a
new phenotype linked to _ATP13A2_. In our UKB cohort, the p.T402M heterozygous mutation carrier was a male PD case with age at recruitment of 55 years. This mutation was absent in controls.
CP The _Ceruloplasmin (CP_) gene, located on chromosome 3, is closely related to iron metabolism in the brain28. Four out of 29 variants had a higher frequency in cases than controls.
Notably, p.R793H and p.P477L, were seen in both AMP-PD and UKB. Reports have linked lower _CP_ levels with PD development, and _CP_ gene mutations have been associated with substantia nigra
hyperechogenicity using transcranial sonography29. In addition, hyperechogenicity is a common finding in idiopathic PD patients and many studies suggest that it is an important risk marker
of future PD30,31. One mutation originally associated with substantia nigra (SN) hyperechogenicity, although listed as likely benign, is p.R793H32. Our study identified the p.R793H variant
in both homozygosity and heterozygosity with higher frequency in cases than controls both AMP-PD and UKB. However, it did not reach statistical significance for either dataset. Of note, we
observed biallelic heterozygosity in one case and two controls from UKB carrying both the p.P477L and p.R793H mutations. The p.I392F variant was only present in UKB data. A three base pair
deletion at this position has been associated with aceruloplasminemia, a disorder in which iron gradually accumulates in the brain and other organs33. In addition, this is a multi-allelic
variant for which the risk allele identified in our analyses differs from those reported on HGMD. FA2H In the _Fatty Acid 2-Hydroxylase_ (_FA2H_) gene, located on chromosome 16, we found 28
coding variants out of which two missense variants had a higher frequency in cases. The variant p.E78K appeared in both datasets and is associated with the fatty acid hydroxylase-associated
neurodegeneration(FAHN)/hereditary spastic paraplegia (SPG35) phenotype although no conclusions were drawn about its significance34. The p.E78K mutation has also been reported in a patient
with an age at onset of 10 with a positive family history of spastic paraplegia and was labeled as a putative pathogenic variant35. Our PD carriers with the p.E78K mutation had an age of
inclusion of 70 and 52 for AMP-PD and 69 for UKB. The case/control frequency for p.E78K did not reach significance in our analysis. For the p.T207M mutation, all previously reported carriers
were compound heterozygous with additional mutations in _FA2H_ and had the spastic paraplegia phenotype22,34,36. In our study, we found one male UKB case with an age at inclusion of 65
carrying the p.T207M mutation. This mutation was absent in controls and was the only _FA2H_ mutation that reached statistical significance. C19ORF12 Variation in the _Chromosome 19 open
reading frame 12_ (_C19orf12_) gene, located on chromosome 19, has been associated with the mitochondrial membrane protein-associated neurodegeneration (MPAN) subtype of NBIA which presents
with cognitive decline progressing to dementia, prominent neuropsychiatric abnormalities, motor neuronopathy, and parkinsonism movement abnormalities37,38. Out of 18 coding variants present,
four missense variants in our datasets showed a higher frequency in cases versus controls, with only one proving significant and previously linked to the MPAN phenotype in the literature.
In AMP-PD data, we found two multiallelic variants, p.G65V present in one case and one control and p.G65A present in just one case and no controls. In two patients reported in the
literature, compound heterozygosity for p.G65V and p.G69RfsX10 variants led to the MPAN phenotype38. In addition, another two patients were reported to have this phenotype; one carrying the
compound heterozygous mutations p.G65V and p.P60L and the other carrying a homozygous p.G65V variant39. The p.K142E variant, present it our data, has been seen in compound heterozygosity in
patients with the MPAN NBIA phenotype40,41,42. Lastly, we found the p.P60L mutation at statistical significance present in one UKB case and absent in controls. The carrier under study was a
female PD patient with an age at inclusion of 66 and no other notable _C19orf12_ mutations. The p.P60L variant has been previously detected in an MPAN patient who also carried the p.G65E
_C19orf12_ mutation38. FTL Our study identified only one missense variant (p.E104) out of nine with a higher frequency in cases than controls in the _Ferritin Light Chain_ (_FTL_) gene,
located on chromosome 19. The p.E104 mutation was only of note in the AMP-PD dataset and was present in one case and one control (F_A = 2.02 × 10−4, F_U = 1.62 × 10−4). The p.E104 variant
has been reported as causing an L-ferritin deficiency phenotype38,43. PANK2 Variation in the _Pantothenate Kinase 2_ (_PANK2_) gene, located on chromosome 20, has been previously associated
with pantothenate kinase-associated neurodegeneration (PKAN), the most common subtype of NBIA44. Clinical symptoms include dystonia, dysarthria, muscular rigidity, poor balance, and
spasticity44,45. We found six out of 68 distinct coding variants in our dataset with a higher frequency in cases. The p.G521R mutation was the only one present in both AMP-PD and UKB. This
variant results in an unstable and inactive PanK2 protein and has been reported as the most common variant in _PANK2_46. It is often found in a compound heterozygous state in individuals
presenting with the PKAN phenotype46,47. In AMP-PD, two PD cases, one male and one female with ages at inclusion of 54 and 67 respectively, carried the p.G521R mutation in heterozygous state
compared to one control (F_A = 4.05 × 10−4, F_U = 4.05 × 10−4). In UKB, three cases, all males with an average age at inclusion of 62 years old, presented with the p.G521R mutation in
heterozygous state compared to five controls (F_A = 1.36 × 10−3, F_U = 4.43 × 10−4). None of the PD p.G521R carriers from either cohort contained this mutation in compound heterozygous
state. PLA2G6 In the _Phospholipase A2 Group VI_ (_PLA2G6_) gene, located on chromosome 22, we found 100 variants of which seven missense variants had a higher frequency in cases versus
controls. These variants have been previously associated with both infantile neuroaxonal dystrophy (INAD) and PD48,49,50,51,52. INAD is a severe progressive psychomotor disorder, which
presents before the third year of life, characterized by the presence of axonal spheroids throughout the central and peripheral nervous system52,53,54. The variant p.M470V is the only
variant out of three associated with the INAD phenotype that was present in both the AMP-PD and UKB cohorts55. In addition, p.A343T has been associated with PD and we identified 77 cases and
86 controls carrying this mutation in AMP-PD (F_A = 1.58 × 10−2, F_U = 1.41 × 10−20) and 29 cases and 130 controls in UKB data (F_A = 1.31 × 10−2, F_U = 1.15 × 10−2)49,55. Of note, one of
the p.A343T AMP-PD case carriers was a female homozygous for the mutation with an age at inclusion of 65. In addition, a heterozygous carrier of p.A343T in the UKB was a PD case and
additionally carried the p.S34L mutation, also associated with PD49,55. This patient had an age at inclusion of 60. However, no _PLA2G6_ mutation with a higher frequency in cases reached
statistical significance. TRANSCRIPTOMIC ANALYSES Two genes including _FA2H_ (ENSG00000103089.8) and _CP_ (ENSG00000047457.13) were removed for further analyses based on poor call rates
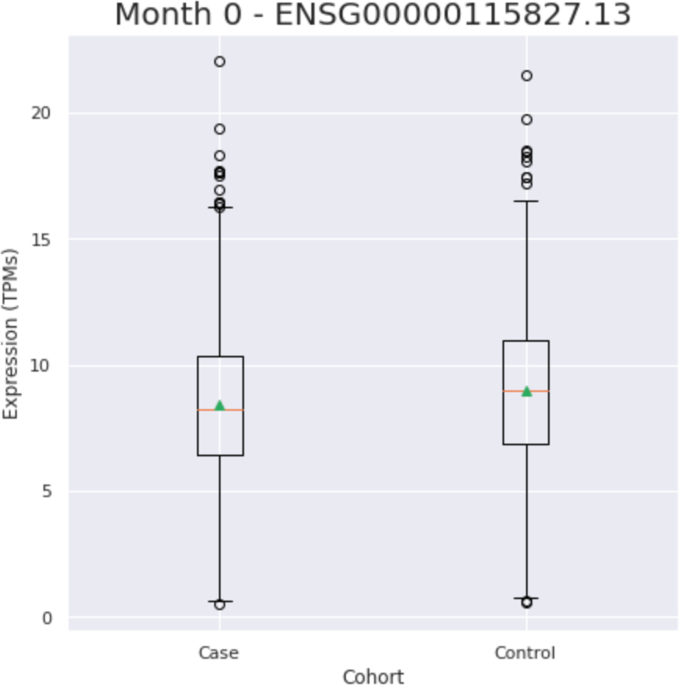
(missingness rate > 0.33). Of the 10 NBIA genes left, _DCAF17_ expression (ENSG00000115827.13) was found to be significantly different in cases versus controls (Fig. 1)(Table 2). To
further investigate these results we looked into SMR analysis for the _DCAF17_ gene. To prioritize SNV candidates, results were initially filtered by chromosome and base pair position for
the _DCAF17_ gene in hg19. Afterwards, the resulting candidates were filtered by the SMR multi p-value at a threshold of _p_ < 0.05. SMR analyses for the _DCAF17_ gene revealed four eQTL
signals with a significant SMR _p_-value. Out of these four eQTL signals, only one was significant in both SMR analyses (_p_ = 0.0024) and Nalls et al.67 GWAS (_p_ = 0.0004). A summary of
the DCAF17 SMR data can be found in the supplementary data. CUMULATIVE EFFECT OF GENETIC VARIATION THROUGH SINGLE GENE AND GENE-SET BURDEN ANALYSES When considering the cumulative effect of
rare variation on PD risk, single gene and gene-set burden analyses did not reveal any statistically significant differences between cases and controls at MAF < 0.01 or MAF < 0.03 in
any of the three cohorts and after meta-analysis (Table 3). DISCUSSION Using the largest PD genetic and genomic datasets available to date, we conducted a comprehensive genetic assessment of
NBIA related variants and genes for a potential role in PD etiology. We identified 29 previously reported NBIA coding mutations in a total of seven genes including _ATP13A2, CP, FA2H,
C19orf12, FTL, PANK2,_ and _PLA2G6_ with a higher frequency in PD cases than controls. All phenotypes previously linked to the reported variants in NBIA genes involve neurological disorders,
adding to the conclusion of clinical overlap between neurological diseases and the strong link between brain iron accumulation and a number of neurodegenerative disorders. However, only
four out of the 29 variants of interest we reported had a statistically significant difference in frequency between cases and controls (_p_ < 0.05), all only significant in UKB data.
Within these four variants, one was found in _ATP13A2_, one in _FA2H_, one in _C19orf12_, and one in _PANK2_, always in only one case and no controls. Notably, none of these four variants
have been previously associated with PD in the literature. Noting the very low mutation frequency in the current data and the lack of replication, we cannot conclude that the mutation
spectrum contributing to NBIA phenotype overlaps with PD etiology. As such, despite enrichment for the risk allele in PD cases versus controls in four variants, these data suggest that in
their heterozygous state NBIA variants are not frequently associated with risk of PD. We performed burden analyses at two different frequency levels, MAF < 0.01 and MAF < 0.03, on both
individual genes and as a gene-set. Meta-analysis of SKAT-O results were not significant for any individual gene or as a gene-set at either frequency level, suggesting that a cumulative
effect of rare variation on NBIA genes does not play a role on PD etiology (Fig. 2). Transcriptomic analyses revealed that _DCAF17_ is the only gene that is differentially expressed in blood
from PD cases and controls. We further investigated this gene through Summary data-based Mendelian Randomization. One _DCAF17_ eQTL signal was significant for both GWAS and SMR. The
_DCAF17_ gene is found on chromosome 2, encodes DDB1- and CUL4- associated factor 17, and is considered the cause of Woodhouse-Sakati syndrome, a rare neuroendocrine disorder that presents
with neurological problems such as seizures and dystonia. While the exact role of the protein is still unclear, it is postulated that it may affect cellular activities such as growth,
proliferation, and stress response specifically in the nucleolus of brain, liver, and skin cells56. It is possible that differing cellular stress responses between PD cases and controls may
be leading to _DCAF17_ differential expression. However, it is still unclear what is driving the significant _DCAF17_ differential gene expression in our data and further assessments are
needed. This study has some limitations, mostly related to variant detection. Despite using the most well-powered PD datasets, it is nevertheless difficult to detect rare variants as
evidenced by the number of variant counts of zero in both cases and controls. It is possible that other rare variants exist in NBIA genes that were not detected in our study. Therefore, we
cannot fully elucidate the relationship between rare variants in NBIA genes and PD. In addition, due to a low number of non-European individuals found in the chosen datasets, we were
underpowered to properly detect variation in other populations. Several the variants we identified have been reported in the literature in non-European populations, so a study encompassing a
greater genetic diversity, with a larger number of non-European individuals, is warranted and would allow us to better characterize the relationship between NBIA and PD etiologies. In
addition, as we have mainly focused on coding variants, it is also possible that non-detected variants, such as larger structural variant (SVs) events in NBIA genes, affect PD risk. There is
increasing evidence that SVs are likely the main contribution to disease susceptibility and have a substantial effect in coding regions of the genome where they can result in deleterious
alterations of the DNA sequence57. Therefore, further study into SVs would be an important step in identifying the relationship between NBIA and PD. Lastly, the cohorts currently available
do not allow us to accurately perform a comprehensive clinical characterization of potential atypical symptoms nor are we able to perform clinicogenetic correlations exploring brain iron
accumulation on MRI in PD cases with NBIA gene variants, therefore possibly drawing limited conclusions. In addition, as it has been previously suggested elevated nigral iron levels are
unlikely to contribute to PD etiology but may vary with anti-parkinsonian drugs used for treatment58. Taking everything into account, we suggest that even though NBIA and PD share similar
symptoms, they could be molecularly different entities supporting the notion that the mechanisms underpinning iron accumulation in PD are not shared with NBIA. Elevated nigral iron levels
may not contribute to PD etiology and may vary with anti-parkinsonian drugs used for treatment or any other environmental factors. METHODS WHOLE-GENOME SEQUENCING DATA Whole-genome
sequencing (WGS) data was obtained from the Accelerating Medicines Partnership—Parkinson’s Disease Initiative release 2.5 (AMP-PD; www.amp-pd.org) which contained 3376 PD patients and 4610
healthy unrelated controls of European ancestry, with an average age at onset of 62 years in cases and age at collection of 72 years in controls. Ancestry was determined by principal
component analysis versus the 1000Genomes populations and individuals deviating by more than six standard deviations from the European population mean were excluded from the analysis.
Diagnosis criteria vary slightly between each subcohort within AMP-PD, but detailed cohort characteristics, as well as quality control procedures, are further described in
https://amp-pd.org/whole-genome-data. Informed consent was obtained for all human participants in this cohort. Healthy unrelated controls include individuals with no neurodegenerative
disease diagnosis nor any family history of such disease lacking any familial link to other individuals in our study. Data generation is described in detail by Iwaki and colleagues59.
NBIA-related variants were extracted from the data by using PLINK1.9 and annotated with ANNOVAR60,61. Coding variants present in the Human Gene Mutation Database (HGMD) went through
association analyses for risk of PD using Fisher’s exact test and logistic regression and adjusted by sex, age, and at least 5PCs to account for population substructure. To assess the
cumulative effect of multiple rare variants on the risk for PD, we performed single gene and gene-set burden analyses for all extracted variants under RVTESTS package v2.1.0 default
parameters and adjusted by sex, age, and at least 5 PCs to account for population substructure62. Including all variants within the gene boundaries, a minimum allele count (MAC) threshold of
1 was applied. Sequence Kernel Association Test (SKAT-O) was performed considering two variant frequency levels: MAF < 1% and MAF < 3%. Age, sex, and at least 5 PCs, were accounted
for as covariates. WHOLE-EXOME SEQUENCING DATA Whole-exome sequencing (WES) data was obtained from the UK Biobank 2021 release (UK Biobank; https://www.ukbiobank.ac.uk/), which contained
1105 PD patients and 5643 healthy unrelated controls of European ancestry, with an average age at recruitment of 63 years in cases and 64 years in controls. Ancestry determination follows
that of AMP-PD. Disease diagnosis of individuals is determined by self-reports and hospital codes and healthy unrelated controls include individuals with no neurodegenerative disease
diagnosis nor any family history of such disease lacking any familial link to other individuals in our study. Detailed cohort characteristics, as well as quality control procedures, are
further described in https://www.ukbiobank.ac.uk/enable-your-research/about-our-data/genetic-data. Informed consent was obtained for all human participants in this cohort. Variant
extraction, association tests, and burden tests follow the AMP-PD WGS pipeline. TRANSCRIPTOMICS DATA Whole blood time progression gene expression data (gencode v29) from 0 to 24 months after
first study visit was accessed from AMP-PD. We used PPMI (https://amp-pd.org/unified-cohorts/ppmi), PDBP (https://amp-pd.org/unified-cohorts/pdbp), and BioFIND
(https://amp-pd.org/unified-cohorts/biofind) cohorts at the baseline of the study, 0 month time point (note BioFIND ‘Baseline’ is at ‘0.5’ month), including a total of 1886 cases and 1285
control samples. Expression data were quantified as transcripts per kilobase million (TPM) and quantile normalized before NBIA related genes values were extracted using Ensembl gene ids.
Using the scikit-learn Python packages 19, residuals were calculated using linear regression, and the data were adjusted using age, sex, race, at least 5 PCs, cohort, and missingness rate as
covariates. To test for significant differences in expression between the cases and controls, we performed a _t_-test with the residuals. We also used an additional dataset taken from the
24-month time point in the PDBP and PPMI cohorts of 841 cases and 386 controls to confirm our findings. SUMMARY BASED MENDELIAN RANDOMIZATION SMR is a mendelian randomization (MR) method
that uses summary-level data to test if an exposure variable (i.e. gene expression) and outcome (i.e., trait) are associated because of a shared causal variant. In order to distinguish
pleiotropy from linkage, the heterogeneity in dependent instruments (HEIDI) method was applied to each tested single nucleotide variant (SNV)63. SMR and HEIDI analysis were conducted using
the SMR package64. In order to conduct the analysis, we used PD GWAS summary statistics as well as methylation and eQTL meta-analysis summary statistics65,66. PD GWAS summary statistics from
Nalls et al.67 contained 17 datasets which consisted of 37,688 PD cases, 18,618 proxy cases, and 1,417,791 controls67. The methylation data we used consists of brain and blood gene
expression data from a meta-analysis conducted by Qi et al.65. All eQTL analyses were performed to test for potential in cis association between methylation site and SNV. In order to achieve
this, SNVs within 1 Mb of the probes of interest were selected65. The eQTL summary statistic data used were generated from peripheral blood and came from the meta-analysis conducted by Wu
et al.68. All of the SNPS from Wu et al. are located within 2 Mb of a probe68. In order to prioritize SNP candidates, results were initially filtered by chromosome and base pair position for
the _DCAF17_ gene in hg19. Afterwards, the resulting candidates were filtered by the SMR multi p-value at a threshold of _p_ < 0.05. REPORTING SUMMARY Further information on research
design is available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY Data used for this publication is available through the AMP-PD and UKBiobank websites
upon request. Data access is dependent on approval of a Data Usage Agreement. AMP-PD Main Website: www.amp-pd.org; AMP-PD Cohort Information: https://amp-pd.org/whole-genome-data; UKBiobank
Main Website: https://www.ukbiobank.ac.uk/; UKBiobabank Cohort Information: https://www.ukbiobank.ac.uk/enable-your-research/about-our-data/genetic-data. CODE AVAILABILITY Code used in this
study can be found at our GitHub page: https://github.com/neurogenetics/NBIA_PD. REFERENCES * Schneider, S. A. Neurodegeneration with brain iron accumulation. _Curr. Neurol. Neurosci. Rep._
16, 9 (2016). Article PubMed Google Scholar * Hogarth, P. Neurodegeneration with brain iron accumulation: diagnosis and management. _J. Mov. Disord._ 8, 1–13 (2015). Article PubMed
PubMed Central Google Scholar * Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R. & Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. _Lancet Neurol._
13, 1045–1060 (2014). Article CAS PubMed PubMed Central Google Scholar * Li, K. R. et al. Quantitative evaluation of brain iron accumulation in different stages of Parkinson’s disease.
_J. Neuroimaging_ 32, 363–371 (2022). Article PubMed Google Scholar * Dexter, D. T. et al. Increased nigral iron content and alterations in other metal ions occurring in brain in
Parkinson’s disease. _J. Neurochem._ 52, 1830–1836 (1989). Article CAS PubMed Google Scholar * Sofic, E., Paulus, W., Jellinger, K., Riederer, P. & Youdim, M. B. Selective increase
of iron in substantia nigra zona compacta of Parkinsonian brains. _J. Neurochem._ 56, 978–982 (1991). Article CAS PubMed Google Scholar * Duran, R. et al. Oxidative stress and
aminopeptidases in Parkinson’s disease patients with and without treatment. _Neurodegener. Dis._ 8, 109–116 (2011). Article CAS PubMed Google Scholar * Agil, A. et al. Plasma lipid
peroxidation in sporadic Parkinson’s disease. Role of the L-dopa. _J. Neurol. Sci._ 240, 31–36 (2006). Article CAS PubMed Google Scholar * Arber, C. E., Li, A., Houlden, H. & Wray,
S. Review: Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories. _Neuropathol. Appl. Neurobiol._ 42, 220–241 (2016). Article
CAS PubMed Google Scholar * Singleton, A. & Hardy, J. A generalizable hypothesis for the genetic architecture of disease: pleomorphic risk loci. _Hum. Mol. Genet._ 20, R158–R162
(2011). Article CAS PubMed PubMed Central Google Scholar * Guerreiro, R. et al. TREM2 variants in Alzheimer’s disease. _N. Engl. J. Med._ 368, 117–127 (2013). Article CAS PubMed
Google Scholar * Jonsson, T. et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. _N. Engl. J. Med._ 368, 107–116 (2013). Article CAS PubMed Google Scholar *
Sidransky, E. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. _N. Engl. J. Med._ 361, 1651–1661 (2009). Article CAS PubMed PubMed Central Google
Scholar * Mochizuki, H. & Yasuda, T. Iron accumulation in Parkinson’s disease. _J. Neural Transm._ 119, 1511–1514 (2012). Article CAS PubMed Google Scholar * Masaldan, S., Bush, A.
I., Devos, D., Rolland, A. S. & Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. _Free Radic. Biol. Med._ 133, 221–233 (2019). Article
CAS PubMed Google Scholar * Carpenter, K. L. H. et al. Magnetic susceptibility of brain iron is associated with childhood spatial IQ. _Neuroimage_ 132, 167–174 (2016). Article CAS
PubMed Google Scholar * Belaidi, A. A. & Bush, A. I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. _J. Neurochem._ 139, 179–197 (2016).
Article CAS PubMed Google Scholar * Barbosa, J. H. O. et al. Quantifying brain iron deposition in patients with Parkinson’s disease using quantitative susceptibility mapping, R2 and R2.
_Magn. Reson. Imaging_ 33, 559–565 (2015). Article CAS PubMed Google Scholar * Hinarejos, I., Machuca-Arellano, C., Sancho, P. & Espinós, C. Mitochondrial dysfunction, oxidative
stress and neuroinflammation in neurodegeneration with brain iron accumulation (NBIA). _Antioxidants_ 9, 1020 (2020). Article CAS PubMed PubMed Central Google Scholar * Wang, Z.-B. et
al. Neurodegeneration with brain iron accumulation: Insights into the mitochondria dysregulation. _Biomed. Pharmacother._ 118, 109068 (2019). Article CAS PubMed Google Scholar * Ramirez,
A. et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. _Nat. Genet._ 38, 1184–1191 (2006). Article CAS PubMed
Google Scholar * Kara, E. et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. _Brain_ 139, 1904–1918 (2016). Article PubMed PubMed Central Google
Scholar * Malakouti-Nejad, M. et al. Identification of p.Gln858* in ATP13A2 in two EOPD patients and presentation of their clinical features. _Neurosci. Lett._ 577, 106–111 (2014). Article
CAS PubMed Google Scholar * Djarmati, A. et al. ATP13A2 variants in early-onset Parkinson’s disease patients and controls. _Mov. Disord._ 24, 2104–2111 (2009). Article PubMed Google
Scholar * Di Fonzo, A. et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. _Neurology_ 68, 1557–1562 (2007). Article PubMed Google Scholar *
Podhajska, A. et al. Common pathogenic effects of missense mutations in the P-type ATPase ATP13A2 (PARK9) associated with early-onset parkinsonism. _PLoS ONE_ 7, e39942 (2012). Article CAS
PubMed PubMed Central Google Scholar * De Michele, G. et al. Ataxia-myoclonus syndrome due to a novel homozygous ATP13A2 mutation. _Parkinsonism Relat. Disord._ 76, 42–43 (2020).
Article PubMed Google Scholar * Hellman, N. E. & Gitlin, J. D. Ceruloplasmin metabolism and function. _Annu. Rev. Nutr._ 22, 439–458 (2002). Article CAS PubMed Google Scholar *
Zhao, X. et al. Ceruloplasmin in Parkinson’s disease and the nonmotor symptoms. _Brain Behav._ 8, e00995 (2018). Article PubMed PubMed Central Google Scholar * Prasuhn, J. et al.
Neuroimaging correlates of substantia nigra hyperechogenicity in Parkinson’s disease. _J. Parkinsons. Dis._ 12, 1191–1200 (2022). Article CAS PubMed Google Scholar * Shafieesabet, A. et
al. Hyperechogenicity of substantia nigra for differential diagnosis of Parkinson’s disease: a meta-analysis. _Parkinsonism Relat. Disord._ 42, 1–11 (2017). Article PubMed Google Scholar
* Hochstrasser, H. et al. Ceruloplasmin gene variations and substantia nigra hyperechogenicity in Parkinson disease. _Neurology_ 63, 1912–1917 (2004). Article CAS PubMed Google Scholar *
Aydemir, S. T., Bulut, O., Ceylaner, S. & Akbostancı, M. C. Aceruloplasminemia presenting with asymmetric chorea due to a novel frameshift mutation. _Mov. Disord. Clin. Pr._ 7, S67–S70
(2020). Google Scholar * Rattay, T. W. et al. FAHN/SPG35: a narrow phenotypic spectrum across disease classifications. _Brain_ 142, 1561–1572 (2019). Article PubMed PubMed Central Google
Scholar * Shakya, S. et al. Whole exome and targeted gene sequencing to detect pathogenic recessive variants in early onset cerebellar ataxia. _Clin. Genet._ 96, 566–574 (2019). Article
CAS PubMed Google Scholar * Magariello, A. et al. Exome sequencing reveals two FA2H mutations in a family with a complicated form of Hereditary Spastic Paraplegia and psychiatric
impairments. _J. Neurol. Sci._ 372, 347–349 (2017). Article CAS PubMed Google Scholar * Hartig, M., Prokisch, H., Meitinger, T. & Klopstock, T. Mitochondrial membrane
protein-associated neurodegeneration (MPAN). _Int. Rev. Neurobiol._ 110, 73–84 (2013). Article CAS PubMed Google Scholar * Hogarth, P. et al. New NBIA subtype: genetic, clinical,
pathologic, and radiographic features of MPAN. _Neurology_ 80, 268–275 (2013). Article PubMed PubMed Central Google Scholar * Gregory, A. et al. Autosomal dominant mitochondrial membrane
protein-associated neurodegeneration (MPAN). _Mol. Genet. Genom. Med._ 7, e00736 (2019). Google Scholar * Hartig, M. B. et al. Absence of an orphan mitochondrial protein, c19orf12, causes
a distinct clinical subtype of neurodegeneration with brain iron accumulation. _Am. J. Hum. Genet._ 89, 543–550 (2011). Article CAS PubMed PubMed Central Google Scholar * Tschentscher,
A. et al. Analysis of the C19orf12 and WDR45 genes in patients with neurodegeneration with brain iron accumulation. _J. Neurol. Sci._ 349, 105–109 (2015). Article CAS PubMed Google
Scholar * Akçakaya, N. H. et al. Clinical and genetic spectrum of an orphan disease MPAN: a series with new variants and a novel phenotype. _Neurol. Neurochir. Pol._ 53, 476–483 (2019).
Article PubMed Google Scholar * Cozzi, A. et al. Human L-ferritin deficiency is characterized by idiopathic generalized seizures and atypical restless leg syndrome. _J. Exp. Med._ 210,
1779–1791 (2013). Article CAS PubMed PubMed Central Google Scholar * Svetel, M. et al. NBIA syndromes: a step forward from the previous knowledge. _Neurol. India_ 69, 1380–1388 (2021).
PubMed Google Scholar * Hayflick, S. J., Kurian, M. A. & Hogarth, P. Neurodegeneration with brain iron accumulation. _Handb. Clin. Neurol._ 147, 293–305 (2018). Article PubMed PubMed
Central Google Scholar * Liang, T.-W. et al. Partial deficit of pantothenate kinase 2 catalytic activity in a case of tremor-predominant neurodegeneration with brain iron accumulation.
_Mov. Disord._ 21, 718–722 (2006). Article PubMed Google Scholar * DaRe, J. T., Vasta, V., Penn, J., Tran, N.-T. B. & Hahn, S. H. Targeted exome sequencing for mitochondrial disorders
reveals high genetic heterogeneity. _BMC Med. Genet._ 14, 118 (2013). Article CAS PubMed PubMed Central Google Scholar * Yeksan, M., Türk, S., Polat, M., Ciğli, A. & Erdoğan, Y.
Effects of 1,25 (OH)2D3 treatment on lipid levels in uremic hemodialysis patients. _Int. J. Artif. Organs_ 15, 704–707 (1992). Article CAS PubMed Google Scholar * Ghani, M. et al.
Mutation analysis of patients with neurodegenerative disorders using NeuroX array. _Neurobiol. Aging_ 36, 545.e9–14 (2015). Article CAS PubMed Google Scholar * Daida, K. et al. PLA2G6
variants associated with the number of affected alleles in Parkinson’s disease in Japan. _Neurobiol. Aging_ 97, 147.e1–147.e9 (2021). Article CAS PubMed Google Scholar * Morgan, N. V. et
al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. _Nat. Genet._ 38, 752–754 (2006). Article CAS PubMed PubMed Central Google
Scholar * Karaca, E. et al. Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. _Neuron_ 88, 499–513 (2015). Article CAS
PubMed PubMed Central Google Scholar * Khateeb, S. et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. _Am. J. Hum. Genet._ 79, 942–948 (2006). Article CAS PubMed PubMed
Central Google Scholar * Iannello, G. et al. A new PLA2G6 mutation in a family with infantile neuroaxonal dystrophy. _J. Neurol. Sci._ 381, 209–212 (2017). Article PubMed Google Scholar
* Arslan, E. A., Öncel, İ., Ceylan, A. C., Topçu, M. & Topaloğlu, H. Genetic and phenotypic features of patients with childhood ataxias diagnosed by next-generation sequencing gene
panel. _Brain Dev._ 42, 6–18 (2020). Article PubMed Google Scholar * Fozia, F. et al. Novel splicing-site mutation in DCAF17 gene causing Woodhouse-Sakati syndrome in a large
consanguineous family. _J. Clin. Lab. Anal._ 36, e24127 (2022). Article CAS PubMed Google Scholar * Pittman, A. & Hardy, J. Genetic analysis in neurology: the next 10 years. _JAMA
Neurol._ 70, 696–702 (2013). Article PubMed PubMed Central Google Scholar * Du, G. et al. Dynamics of nigral iron accumulation in Parkinson’s disease: from diagnosis to late stage. _Mov.
Disord._ 37, 1654–1662 (2022). Article CAS PubMed PubMed Central Google Scholar * Iwaki, H. et al. Accelerating medicines partnership: Parkinson’s disease. genetic resource. _Mov.
Disord._ 36, 1795–1804 (2021). Article CAS PubMed PubMed Central Google Scholar * Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets.
_GigaScience_ 4 (2015). * Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. _Nucleic Acids Res._ 38, e164 (2010).
Article PubMed PubMed Central Google Scholar * Zhan, X., Hu, Y., Li, B., Abecasis, G. R. & Liu, D. J. RVTESTS: an efficient and comprehensive tool for rare variant association
analysis using sequence data. _Bioinformatics_ 32, 1423–1426 (2016). Article CAS PubMed PubMed Central Google Scholar * Zhu, Z. et al. Integration of summary data from GWAS and eQTL
studies predicts complex trait gene targets. _Nat. Genet._ 48, 481–487 (2016). Article CAS PubMed Google Scholar * Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies
predicts complex trait gene targets. _Nat. Genet._ 48, 481–487 (2016). Article CAS PubMed Google Scholar * Qi, T. et al. Identifying gene targets for brain-related traits using
transcriptomic and methylomic data from blood. _Nat. Commun._ 9, 2282 (2018). Article PubMed PubMed Central Google Scholar * McRae, A. F. et al. Identification of 55,000 replicated DNA
methylation QTL. _Sci. Rep._ 8, 17605 (2018). Article PubMed PubMed Central Google Scholar * Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk
for Parkinson’s disease: a meta-analysis of genome-wide association studies. _Lancet Neurol._ 18, 1091–1102 (2019). Article CAS PubMed PubMed Central Google Scholar * Wu, Y. et al.
Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. _Nat. Commun._ 9, 918 (2018). Article PubMed PubMed Central Google Scholar * Adapted
from “Flowchart 5”, by BioRender.com. Retrieved from https://app.biorender.com/biorender-templates (2023). Download references ACKNOWLEDGEMENTS Data used in the preparation of this article
were obtained from Global Parkinson’s Genetics Program (GP2). GP2 is funded by the Aligning Science Against Parkinson’s (ASAP) initiative and implemented by The Michael J. Fox Foundation for
Parkinson’s Research (https://gp2.org). For a complete list of GP2 members see https://gp2.org. The work was carried out with the support and guidance of the ‘GP2 Trainee Network’. This
work was supported by the Center for Alzheimer’s and Related Dementias, within the Intramural Research Program of the National Institute on Aging and the National Institute of Neurological
Disorders and Stroke, National Institutes of Health, Department of Health and Human Services. Project number 1ZIA AG000534-04. AMP-PD: Data used in the preparation of this article were
partly obtained from the Accelerating Medicine Partnership® (AMP®) Parkinson’s Disease (AMP PD) Knowledge Platform. For up-to-date information on the study, visit https://www.amp-pd.org. The
AMP® PD program is a public-private partnership managed by the Foundation for the National Institutes of Health and funded by the National Institute of Neurological Disorders and Stroke
(NINDS) in partnership with the Aligning Science Across Parkinson’s (ASAP) initiative; Celgene Corporation, a subsidiary of Bristol-Myers Squibb Company; GlaxoSmithKline plc (GSK); The
Michael J. Fox Foundation for Parkinson’s Research; Pfizer Inc.; Sanofi US Services Inc.; and Verily Life Sciences. ACCELERATING MEDICINES PARTNERSHIP and AMP are registered service marks of
the U.S. Department of Health and Human Services. Clinical data and biosamples used in preparation of this article were obtained from the Michael J. Fox Foundation (MJFF) and National
Institutes of Neurological Disorders and Stroke (NINDS) BioFIND study, Harvard Biomarkers Study (HBS), the NINDS Parkinson’s disease Biomarkers Program (PDBP) and MJFF Parkinson’s
Progression Marker Initiative (PPMI). BioFIND is sponsored by The Michael J. Fox Foundation for Parkinson’s Research (MJFF) with support from the National Institute for Neurological
Disorders and Stroke (NINDS). The BioFIND Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit
michaeljfox.org/news/biofind. The Harvard NeuroDiscovery Biomarker Study (HBS) is a collaboration of HBS investigators [full list of HBS investigator found at
https://www.bwhparkinsoncenter.org/biobank/ and funded through philanthropy and NIH and Non-NIH funding sources. The HBS Investigators have not participated in reviewing the data analysis or
content of the manuscript. PPM—a public-private partnership – is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, a full list of the PPMI funding
partners can be found at www.ppmi-info.org/fundingpartners. The PPMI Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date
information on the study, visit www.ppmi-info.org. Parkinson’s Disease Biomarker Program (PDBP) consortium is supported by the National Institute of Neurological Disorders and Stroke (NINDS)
at the National Institutes of Health. A full list of PDBP investigators can be found at https://pdbp.ninds.nih.gov/policy. The PDBP Investigators have not participated in reviewing the data
analysis or content of the manuscript. UKB: UK Biobank is a large-scale biomedical database and research resource containing genetic, lifestyle and health information from half a million UK
participants. UK Biobank’s database, which includes blood samples, heart and brain scans and genetic data of the 500,000 volunteer participants, is globally accessible to approved
researchers who are undertaking health-related research that’s in the public interest. UK Biobank recruited 500,000 people aged between 40–69 years in 2006–2010 from across the UK. With
their consent, they provided detailed information about their lifestyle, physical measures and had blood, urine and saliva samples collected and stored for future analysis. UK Biobank’s
research resource is a major contributor in the advancement of modern medicine and treatment, enabling better understanding of the prevention, diagnosis and treatment of a wide range of
serious and life-threatening illnesses—including cancer, heart diseases and stroke. UK Biobank is generously supported by its founding funders the Wellcome Trust and UK Medical Research
Council, as well as the Department of Health, Scottish Government, the Northwest Regional Development Agency, British Heart Foundation and Cancer Research UK. The organization has over 150
dedicated members of staff, based in multiple locations across the UK. 23andme: We thank the 23andMe research participants who consented to participate in research for enabling this study.
We also thank the employees of 23andMe. FUNDING Open Access funding provided by the National Institutes of Health (NIH). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Molecular Genetics
Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, MD, USA Pilar Alvarez Jerez, Anni Moore, Mary B. Makarious, Kimberley J.
Billingsley, Cornelis Blauwendraat, Andrew B. Singleton & Sara Bandres-Ciga * Center for Alzheimer’s and Related Dementias (CARD), National Institute on Aging and National Institute of
Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, USA Pilar Alvarez Jerez, Chelsea X. Alvarado, Kimberley J. Billingsley, Cornelis Blauwendraat, Andrew B.
Singleton & Sara Bandres-Ciga * Department of Neurodegenerative Disease, UCL Queen Square Institute of Neurology, University College London, London, UK Pilar Alvarez Jerez * Institute of
Neurosciences “Federico Olóriz”, Centro de Investigación Biomédica, Universidad de Granada, Granada, Spain Jose Luis Alcantud, Clara Ruz, Francisco Vives Montero & Raquel Duran *
Laboratory of Neuropharmacology. Dept. Medicine and Life Sciences, Universitat Pompeu Fabra, Barcelona, Spain Lucia de los Reyes-Ramírez * Department Human Physiology, Faculty of Medicine,
Biomedicine Research Institute of Malaga (IBIMA C07), University of Malaga, Malaga, Spain Noela Rodriguez-Losada * Montreal Neurological Institute, McGill University, Montréal, QC, Canada
Prabhjyot Saini & Ziv Gan-Or * Department of Human Genetics, McGill University, Montréal, QC, Canada Prabhjyot Saini & Ziv Gan-Or * Department of Neurology and Neurosurgery, McGill
University, Montréal, QC, Canada Ziv Gan-Or * Data Tecnica International, Washington, DC, USA Chelsea X. Alvarado * Department of Clinical and Movement Neurosciences, UCL Queen Square
Institute of Neurology, London, UK Mary B. Makarious & Alastair J. Noyce * Preventive Neurology Unit, Centre for Prevention, Detection and Diagnosis, Wolfson Institute of Population
Health, Queen Mary University of London, London, UK Alastair J. Noyce Authors * Pilar Alvarez Jerez View author publications You can also search for this author inPubMed Google Scholar *
Jose Luis Alcantud View author publications You can also search for this author inPubMed Google Scholar * Lucia de los Reyes-Ramírez View author publications You can also search for this
author inPubMed Google Scholar * Anni Moore View author publications You can also search for this author inPubMed Google Scholar * Clara Ruz View author publications You can also search for
this author inPubMed Google Scholar * Francisco Vives Montero View author publications You can also search for this author inPubMed Google Scholar * Noela Rodriguez-Losada View author
publications You can also search for this author inPubMed Google Scholar * Prabhjyot Saini View author publications You can also search for this author inPubMed Google Scholar * Ziv Gan-Or
View author publications You can also search for this author inPubMed Google Scholar * Chelsea X. Alvarado View author publications You can also search for this author inPubMed Google
Scholar * Mary B. Makarious View author publications You can also search for this author inPubMed Google Scholar * Kimberley J. Billingsley View author publications You can also search for
this author inPubMed Google Scholar * Cornelis Blauwendraat View author publications You can also search for this author inPubMed Google Scholar * Alastair J. Noyce View author publications
You can also search for this author inPubMed Google Scholar * Andrew B. Singleton View author publications You can also search for this author inPubMed Google Scholar * Raquel Duran View
author publications You can also search for this author inPubMed Google Scholar * Sara Bandres-Ciga View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS P.A.J., S.B.C., and R.D. designed the study and wrote the manuscript with inputs from all co-authors. P.A.J. analyzed the data and prepared the figures with supervision from
S.B.C. C.X.A. performed the SMR analysis. A.M. performed the transcriptomic analysis. J.L.A. aided with initial study design and manuscript editing. M.B.M. provided meta-analysis scripts
used and aided in manuscript editing. L.R.M., C.R., F.V.M., N.R.L., and P.S. aided in manuscript writing and editing. Z.G.O., K.J.B., C.B., A.J.N., and A.B.S. provided design supervision and
aided in manuscript editing. All authors approved the final version of the manuscript. All authors take accountability for all aspects of the work. CORRESPONDING AUTHOR Correspondence to
Sara Bandres-Ciga. ETHICS DECLARATIONS COMPETING INTERESTS C.R. and A.B.S. declare no Competing Non-Financial Interests but the following Competing Financial Interests: C.R. held a
predoctoral fellowship (FPU14/03473, MECD, Spain) from the Spanish Ministry of Education and Science. A.B.S. is an editor for _npj Parkinson’s Disease_. A.B.S. was not involved in the
journal’s review of, or decisions related to, this manuscript. P.A.J., J.L.A., L.R.R., A.M., F.V.M., N.R.L., P.S., Z.G.O., C.X.A., M.B.M., K.J.B., C.B., A.J.N., A.B.S., R.D., and S.B.C.
declare no competing interests. ETHICAL APPROVAL We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this work is consistent with
those guidelines. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY TABLES REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Alvarez Jerez, P., Alcantud, J.L., de los Reyes-Ramírez, L. _et al._ Exploring the genetic and genomic connection underlying
neurodegeneration with brain iron accumulation and the risk for Parkinson’s disease. _npj Parkinsons Dis._ 9, 54 (2023). https://doi.org/10.1038/s41531-023-00496-y Download citation *
Received: 14 December 2022 * Accepted: 16 March 2023 * Published: 06 April 2023 * DOI: https://doi.org/10.1038/s41531-023-00496-y SHARE THIS ARTICLE Anyone you share the following link with
will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative
Trending News
Funds could face market curbs after lobbying backfiresFund managers may face tougher scrutiny by global regulators than planned after their intense lobbying against a first p...
Page Not Found - GSMArena.comGSMArena.com Tip us 2.0m 150k RSS EV Merch Log in LoginI forgot my password Sign up Home News Reviews Videos Featured Ph...
Never! Never!_Por Percival Puggina_ “O Destino de uma Nação” chegou aos cinemas suscitando muitas abordagens na imprensa brasileira....
Leavers are the ones who should be demanding a second referendum | thearticleHow often do we hear exclaimed “This is not the Brexit I voted for” to pre-empt some particular Brexit arrangement the g...
The end of a great French culinary institution | TheArticleCulture and Civilisations Saturday November 30, 2019 The end of a great French culinary institution by Giles MacDonogh| ...
Latests News
Exploring the genetic and genomic connection underlying neurodegeneration with brain iron accumulation and the risk for parkinson’s diseaseABSTRACT Neurodegeneration with brain iron accumulation (NBIA) represents a group of neurodegenerative disorders charact...
National id contract and the pa consulting groupFOI release NATIONAL ID CONTRACT AND THE PA CONSULTING GROUP FOI 1401 We received a request under the Freedom of Informa...
Amanda holden suffers embarrassing nip slip in plunging white suitAs she slipped into a waiting car, her best assets fell out of her jacket giving onlookers outside an eyeful. Amanda pa...
Ms k herriot v united kingdom independence party ltd: 1402999/2020MS K HERRIOT V UNITED KINGDOM INDEPENDENCE PARTY LTD: 1402999/2020 Employment Tribunal decision. Read the full decision ...
Krispy kreme and pop-tarts plan trio of ‘unexpected’ mashups: ‘crazy good’You _do-nut _want to miss this. Krispy Kreme announced a first-of-its-kind collaboration with Pop-Tarts, which will intr...