Enhanced tlr-induced nf-κb signaling and type i interferon responses in nlrc5 deficient mice
Enhanced tlr-induced nf-κb signaling and type i interferon responses in nlrc5 deficient mice"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Nod-like receptors (NLRs) are intracellular sensors that respond to a variety of pathogen and intracellular danger signals to induce innate immune responses. NLRC5 has recently been
identified to be an important regulator of NF-κB, type I interferon (IFN) and inflammasome signaling pathways, but the _in vivo_ function and mechanisms of NLRC5 remain to be defined. Here,
we describe the generation and characterization of _NLRC5_ knockout mice. We show that induction of _NLRC5_ expression by Toll-like receptor (TLR) ligand or cytokine stimulation requires
the signal transducers and activators of transcription (Stat)1-mediated signaling pathway. _NLRC5_ ablation reduces MHC class I expression, and enhances IKK and IRF3 phosphorylation in
response to TLR stimulation or viral infection. Consistent with these observations, we found that _NLRC5_ deficiency enhanced IL-6 and IFN-β production in mouse embryonic fibroblasts (MEFs),
peritoneal macrophages and bone marrow-derived macrophages (BMMs), but not bone marrow-derived dendritic cells (BMDCs) after LPS stimulation or vesicular stomatitis virus (VSV) infection.
Furthermore, we found that _NLRC5_-deficient mice produced higher amounts of IL-6 and IFN-β in the sera when they were challenged with LPS or infected with VSV. Taken together, these results
provide _in vivo_ evidence that NLRC5 plays critical roles in MHC class I expression, innate immune signaling and antiviral innate immune responses, thus serving as an important target for
modulating innate immune signaling and regulation. SIMILAR CONTENT BEING VIEWED BY OTHERS IFITM3 RESTRICTS VIRUS-INDUCED INFLAMMATORY CYTOKINE PRODUCTION BY LIMITING NOGO-B MEDIATED TLR
RESPONSES Article Open access 08 September 2022 INTEGRATION OF INNATE IMMUNE SIGNALLING BY CASPASE-8 CLEAVAGE OF N4BP1 Article 24 September 2020 THE TLR7/9 ADAPTORS TASL AND TASL2 MEDIATE
IRF5-DEPENDENT ANTIVIRAL RESPONSES AND AUTOIMMUNITY IN MOUSE Article Open access 24 January 2025 INTRODUCTION The innate immune system is critically important as the first line of defense
against invading pathogens by detecting or sensing pathogen- and danger-associated molecular patterns (known as PAMPs and DAMPs, respectively) through germline-encoded pattern recognition
receptors (PRRs), which include Toll-like receptor (TLR) family, retinoic acid inducible gene I (RIG-I)-like receptor (RLR) family, Nod-like receptor family and DNA sensors1,2,3,4. Upon PAMP
stimulation, these PRRs trigger the activation of NF-κB, type I interferon (IFN) or inflammasome signaling pathways, which leads to the production of proinflammatory and antiviral
cytokines, and induction of subsequent adaptive immune responses against invading pathogens. Activation of most TLRs leads to the recruitment of a common adaptor, MyD88, and a series of
downstream signaling events that culminate in NF-κB activation and inflammatory responses. TLR3 recognizes viral dsRNA in endosomes and activates both NF-κB and type I IFN signaling pathways
through TIR domain-containing adaptor-inducing interferon-β (TRIF). RIG-I and MDA5 (melanoma differentiation-associated gene 5) function as cytoplasmic RNA sensors and recruit the
mitochondrial protein called MAVS (also known as VISA, IPS-1 and Cardif) upon detection of intracellular RNA viruses1,2,3. Interferon-γ inducible protein 16 (IFI16) and DEAD
(Asp-Glu-Ala-Asp) box protein 41 (DDX41) have been identified as cytosolic viral DNA sensors that activate the type I IFN signaling pathway5,6,7,8. NLRs represent a large group of protein
family containing a conserved central nucleotide-binding and oligomerization domain (NOD), a leucine-rich repeat (LRR) region and a variable N-terminal effector domain9. While some NLRs,
such as NOD1, NOD2 and NLRP3, serve as the PRRs in response to various PAMPs, others may function as negative regulators1,2,10. For example, NLRX1 has been shown to inhibit type I IFN
signaling and NF-κB activation by interaction with MAVS or IKKα/IKKβ11,12,13. It is also implicated in the generation of reactive oxygen species14. NLRP4 has been reported to negatively
regulate autophagic processes, NF-κB and type I IFN signaling by interaction with Beclin, IKKβ and TBK1, respectively15,16. More recently, NLRC5 has been identified as an important regulator
of both innate and adaptive immune regulation17,18,19,20,21,22. While two groups have shown that NLRC5 inhibits NF-κB and type I IFN signaling17,18, two other reports indicate that NLRC5 is
required for IFN production in antiviral responses19,20. Furthermore, NLRC5 has been demonstrated to associate with NLRP3 to cooperatively activate the inflammasome21. NLRC5 has also been
identified as a critical regulator of MHC class I gene expression22. Thus, it appears that NLRC5 plays diverse roles in innate immune regulation and MHC class I expression. To determine the
_in vivo_ function of NLRC5, Kumar _et al_.23 generated _NLRC5_-deficient mice by deleting exon 4 of _NLRC5_, but they did not observe any effect of _NLRC5_ ablation on proinflammatory
cytokine production in response to virus and bacteria infection in bone marrow-derived dendritic cells (BMDCs). In this report, we describe the generation of _NLRC5_-deficient mice by
deleting exon 8 of _NLRC5_, and show that _NLRC5_ ablation enhanced NF-κB and type I IFN signaling pathways in response to various TLR stimulation or vesicular stomatitis virus (VSV)
infection in multiple cell types, including mouse embryonic fibroblasts (MEFs), peritoneal macrophages and bone marrow-derived macrophages (BMMs), but not BMDCs. Furthermore,
_NLRC5_-deficient mice produced higher amounts of IL-6 and IFN-β in the sera when they were challenged with LPS or infected with VSV, respectively. Taken together, these results demonstrate
the critical role of NLRC5 in TLR-induced NF-κB signaling and antiviral innate immune responses. RESULTS SIGNAL TRANSDUCERS AND ACTIVATORS OF TRANSCRIPTION (STAT)1 IS A KEY FACTOR FOR NLRC5
INDUCTION Although several groups have shown that NLRC5 can be induced by treatment with TLR ligands such as LPS, polyinosinic-polycytidylic acid (poly(I:C)), cytokines such as IFN-γ and
IFN-β, or virus infection, the mechanisms controlling its induction remain unclear17,18,19,20. To further determine whether NLRC5 induction is regulated by NF-κB or IFN-mediated signaling
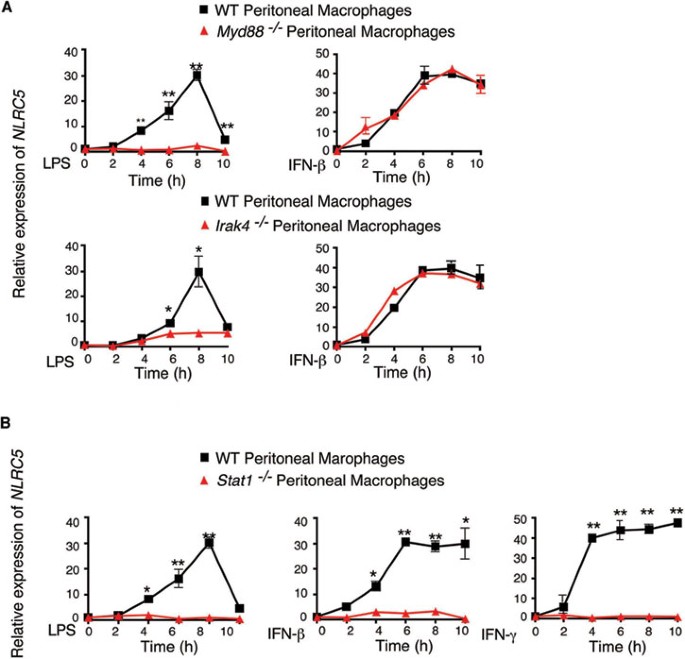
pathway, we assessed _NLRC5_ expression in _MyD88_−/−, _IRAK4_−/− or _Stat1_−/− mouse peritoneal macrophages in response to LPS or IFN-β stimulation. _NLRC5_ expression was abrogated in
_MyD88_−/− or _IRAK4_−/− macrophages after LPS treatment (Figure 1A), consistent with our previous report17. However, induction of _NLRC5_ expression in _MyD88_−/− or _IRAK4_−/− cells was
identical to that in wild-type (WT) cells when they were treated with IFN-β, suggesting that induction of _NLRC5_ expression by IFN-β treatment remained intact in _MyD88_−/− or _IRAK4_−/−
cells and is independent of NF-κB signaling. It is known that the Stat pathway plays a critical role in the expression of many cytokine-induced genes24,25. Since LPS treatment can induce
production of multiple cytokines, including IFN-β and IFN-γ, we reasoned that induction of _NLRC5_ expression by TLR ligands might result from the production of LPS-activated
NF-κB-responsive cytokines, which in turn induce _NLRC5_ expression. To test this possibility, we treated Stat1−/− peritoneal macrophages with LPS, IFN-γ or IFN-β, and found that LPS-, IFN-γ
or IFN-β-mediated _NLRC5_ induction was completely abrogated in Stat1−/− peritoneal macrophages (Figure 1B). These data suggest that _NLRC5_ expression induced by LPS is dependent on Stat1
signaling. Furthermore, we found that the cell supernatants of LPS-treated RAW264.7 cells were sufficient to induce _NLRC5_ expression (Supplementary information, Figure S1A and S1B). IFN-β
neutralizing antibody could almost completely abrogate LPS-mediated _NLRC5_ induction, while IFN-γ neutralizing antibody had little effect (Supplementary information, Figure S1C and S1D),
probably due to very low level of secreted IFN-γ (data not shown). These results suggest that LPS-mediated NF-κB activation leads to the production of IFN-β, which in turn induce _NLRC5_
expression through the Stat1 signaling pathway. NLRC5 DELETION REDUCES MHC CLASS I EXPRESSION IN T CELLS To elucidate the physiological roles of NLRC5, we generated _NLRC5_-deficient
(_NLRC5_−/−) mice by homologous recombination in embryonic stem (ES) cells (Figure 2A and 2B). The targeting vector was constructed by replacing a 2.1-kb fragment, which encodes a functional
LRR1 domain of NLRC5, with a Neo expression cassette (Figure 2A). The linearized targeting vector was then injected into ES cells by electroporation. Homologous recombinant stem cells were
identified by screening with PCR and further confirmed by Southern blotting analysis (Supplementary information, Figure S2A and Figure 2C). The resultant recombinant products led to
termination of NLRC5 open reading frame. The selected homologous recombinant ES clones were injected into blastocysts. Chimeric mice generated from two homologous ES clones were bred to
generate F1 heterozygotes as well as homozygous _NLRC5_-deficient mice. _NLRC5_ knockout mice were born at a Mendelian ratio (Figure 2D) and appeared to be normal, compared with WT mice. A
recent study shows that overexpression of NLRC5 results in enhanced MHC class I expression in lymphoid as well as epithelial cell lines22. To assess the regulatory role of NLRC5 in MHC class
I gene expression _in vivo_, we examined the expression of the mouse MHC class I molecule (H2-Kb) in CD4+ T cells, CD8+ T cells and B cells isolated from _NLRC5_-deficient and WT mice. We
found marked reduction of H2-Kb in _NLRC5_-deficient T cells, compared to WT cells (Figure 2E). We also observed a small difference in MHC class I expression in _NLRC5_-deficient B cells
compared with WT cells (Figure 2E). In contrast, we observed little or no difference in MHC class II expression in _NLRC5_-deficient T cells and B cells (Figure 2E). It should be noted that
MHC class II expression in _NLRC5_-deficient CD8+ T cells was a little higher than that in WT cells. These results suggest that NLRC5 is an important transcriptional regulator of MHC class I
gene expression. NLRC5 ABLATION ENHANCES TLR-INDUCED NF-ΚB ACTIVATION AND PROINFLAMMATORY CYTOKINE PRODUCTION IN MEFS To investigate the role of NLRC5 in the NF-κB pathway under
physiological conditions, we treated MEFs generated from WT and _NLRC5_-deficient mice with LPS, followed by immunoblot analysis of phosphorylation of IKK and MAP kinases (p38 and JNK). We
found that phosphorylation of IKK was markedly enhanced in _NLRC5_−/− MEFs compared with WT MEFs after LPS stimulation. By contrast, we did not observe an appreciable difference in p38 and
JNK phosphorylation between WT and _NLRC5_−/− MEFs (Figure 3A). To determine whether increased NF-κB activation in _NLRC5_−/− cells correlates with expression of NF-κB-responsive cytokine
genes, we treated MEFs with LPS and then analyzed the expression of cytokines. The expression of _IL-6_, _TNF-α_ and _IL-1β_ in _NLRC5_−/− MEFs was significantly higher than in WT MEFs post
LPS infection, but the mRNA level of _Ifn-β_, which is activated through the TLR4-TRIF pathway, was comparable between WT and _NLRC5_−/− cells (Figure 3B). These results are consistent with
data previously reported by our group and others17,18. Collectively, these results suggest that _NLRC5_ deletion enhances NF-κB activation as well as proinflammatory cytokine gene expression
in MEFs following LPS stimulation. We next determined the effect of _NLRC5_ deletion on IL-6 and TNF-α production in MEFs following stimulation with TLR ligands, including poly(I:C), LPS,
CL-097 and CpG, which activate TLR3-, TLR4-, TLR7- and TLR9-mediated signaling pathways, respectively. We observed marked increases in IL-6 in _NLRC5_−/− MEFs compared to WT MEFs after
stimulation with different TLR ligands (Figure 3C). TNF-α production was also increased in _NLRC5_−/− MEFs treated with LPS and CpG, but not with poly(I:C), compared to that in WT MEFs
(Supplementary information, Figure S3). Taken together, these results suggest that _NLRC5_ deletion enhances NF-κB activation and proinflammatory cytokine production in response to multiple
TLR stimulation in MEFs. NLRC5 DEFICIENCY ENHANCES ANTIVIRAL IMMUNITY AGAINST RNA VIRUSES IN MEFS To evaluate the effect of _NLRC5_ deficiency on antiviral innate immunity, we infected MEFs
with a RNA virus, VSV-eGFP. IRF3 was phosphorylated as early as 4 h postinfection with VSV, and gradually increased with time. We found that the phosphorylation of IRF3 was at least twofold
higher in _NLRC5_−/− MEFs, compared to that in WT MEFs by 8 h after viral infection (Figure 4A), while total IRF3 expression was comparable between the two groups. Moreover, loss of _NLRC5_
also increased the expression of type I IFN-responsive genes, including _IFN-α_ and _IFN-β_ after VSV-eGFP infection (Figure 4B). However, little or no differences in _Tnf-α_ and _Il-6_
expression were detected (Figure 4B), consistent with our previous findings obtained from _NLRC5_-specific siRNA knockdown experiments17. ELISA analyses showed that intracellular poly(I:C)
treatment (ligand for MDA5) or VSV-eGFP infection (ligand for RIG-I) strongly increased the secretion of IFN-β in _NLRC5_−/− MEFs compared with that in WT MEFs (Figure 4C). These results
suggest that _NLRC5_ deficiency enhanced IFN-β activation through the RIG-I/MDA5 pathways. To demonstrate a link between increased type I IFN response and antiviral immunity in _NLRC5_−/−
MEFs, we infected the _NLRC5_−/−, WT and _MAVS_−/− cells with VSV-eGFP (MOI = 1), and infection was monitored based on GFP expression. GFP-positive (VSV-eGFP infected) cells could be
detected at as early as 15 h, peaked at 25 h and slightly decreased (due to cell apoptosis) at 30-35 h post-infection in _MAVS_−/− cells (that were defective in type I IFN signaling) (Figure
4D). By contrast, we did not observe GFP-positive (VSV-eGFP infected) cells at 25 h in _NLRC5_−/− cells. The GFP-positive cells became detectable at 30 h and increased slightly at 35 h
postinfection in _NLRC5_−/− cells (Figure 4D). WT MEFs showed intermediate GFP-positive cells (detectable at 20 h and gradually increased with time until 35 h postinfection; Figure 4D).
Thus, _MAVS_−/− cells are more sensitive to VSV-eGFP infection than WT MEFs, while _NLRC5_-deficient MEFs are more resistant to VSV-eGFP infection than WT cells, indicating that _NLRC5_
ablation increases the ability of MEFs to inhibit VSV infection, thus enhancing antiviral immunity. NLRC5 NEGATIVELY REGULATES NF-ΚB AND TYPE I IFN PATHWAYS IN MACROPHAGES To determine
whether NLRC5 negatively regulates NF-κB and type I IFN pathways in immune cells, we performed similar experiments with BMDCs and found little or no difference in IL-6, TNF-α and IL-12
secretion in _NLRC5_−/− and WT BMDCs stimulated with various TLR ligands (Supplementary information, Figure S4A and S4B), which was consistent with a previous report showing that _NLRC5_
deficiency does not affect proinflammatory and type I IFN cytokines production in BMDCs and peritoneal exudates cells after stimulation with TLR agonists and RLRs23. However, when we treated
peritoneal macrophages with LPS, we found that the phosphorylation of IKK was significantly increased in _NLRC5_−/− peritoneal macrophages, while the activation of MAP kinases (p38, ERK and
JNK) was comparable between WT and _NLRC5_−/− peritoneal macrophages (Figure 5A). Furthermore, the mRNA level of _IL-6_ was much higher, while the expression of _TNF-α_ and _IL-1β_ was
increased at early time points in _NLRC5_−/− peritoneal macrophages. The induction of IFN-β was comparable between _NLRC5_−/− and WT cells (Figure 5B). These results indicate that unlike in
BMDCs, NLRC5 plays a negative regulatory role in NF-κB signaling in murine peritoneal macrophages. To further confirm the function of NLRC5 in macrophages, we treated peritoneal macrophages
with different TLR ligands and measured cytokine release. We observed significantly higher release of IL-6, but not TNF-α, in response to stimulation with TLR ligands from 12 to 24 h
poststimulation in _NLRC5_−/− peritoneal macrophages, compared to WT cells (Figure 5C and Supplementary information, Figure S5A). Therefore, NLRC5 plays a negative role in regulation of
NF-κB signaling in peritoneal macrophages. The difference between BMDCs and peritoneal macrophages may be due to the different expression level of NLRC5 (Supplementary information, Figure
S4E). Interestingly, we did not observe any difference in IL-1β secretion between _NLRC5_−/− and WT peritoneal macrophages (Supplementary information, Figure S5B), which is consistent with
another report23. It should be noted that it appears that loss of _NLRC5_ may enhance more proinflammatory cytokine release at low doses of stimulation, as large differences in IL-6 and
TNF-α production were observed between _NLRC5_-deficient and WT cells when a lower dose of LPS (10 ng/ml) was used (Supplementary information, Figure S6). To elucidate the role of NLRC5 in
type I IFN signaling in peritoneal macrophages, we infected cells with VSV-eGFP, and found that IRF3 phosphorylation occurred by 4 h postinfection and gradually increased with time in
_NLRC5_−/− cells. However, IRF3 phosphorylation was barely detectable until 8 h postinfection in WT macrophages, and p-IRF3 levels in WT cells were generally lower than in _NLRC5_−/− cells
(Figure 5D). In addition, VSV-eGFP-infected (GFP-positive) cells in _NLRC5_−/− macrophages were much less than in WT cells (Figure 5E), suggesting that _NLRC5_−/− macrophages show enhanced
antiviral immunity compared to WT cells. To further investigate the specificity of the inhibitory function of NLRC5 in type I IFN signaling, we treated peritoneal macrophages with VSV-eGFP,
poly(I:C)/LyoVec (intracellular poly(I:C)), poly(I:C) and LPS to activate RIG-I-, MDA5-, TLR3- and TLR4-meidated type I IFN signaling, respectively. IFN-β secretion was highly increased in
_NLRC5_−/− cells in response to RIG-I/MDA5 ligands (VSV or intracellular poly(I:C)), but was barely changed when the cells were stimulated with LPS or poly(I:C) (Figure 5F), consistent with
our previous findings that NLRC5 mainly interacts with RIG-I/MDA5 to block RNA virus-induced type I IFN signaling17. To further confirm the role of NLRC5 in immune response to TLR ligands or
virus infection in macrophages, we isolated BMMs and treated the cells with TLR ligands or infected them with VSV or dsRNA. We observed a similar pattern of IKK and MAP kinase activation in
_NLRC5_−/− BMMs to that in peritoneal macrophages (Figure 6A). We also observed that the secretion of IL-6, but not TNF-α, was higher in NRLC5−/− BMMs compared with WT BMMs, but this
difference was not as dramatic as observed in peritoneal macrophages (Figure 6B and Supplementary information, Figure S5C). In addition, we found that phosphorylation of IRF3 was increased
after VSV-eGFP infection in _NLRC5_−/− BMMs compared to WT BMMs (Figure 6C). To further support that NLRC5 inhibits antiviral responses through RIG-I/MDA5 pathways in macrophages, we
evaluated the antiviral response after VSV-eGFP, poly(I:C)/LyoVec or poly(I:C) treatment by measuring the IFN-β release in culture supernatants. _NLRC5_−/− BMMs produced more IFN-β in
response to VSV or poly(I:C)/LyoVec, but not poly(I:C), compared to WT BMMs (Figure 6D). Collectively, these results suggest that _NLRC5_ ablation enhances both NF-κB and type I interferon
pathways in macrophages. THE FUNCTION OF NLRC5 IN INFLAMMATION AND ANTIVIRAL IMMUNITY _IN VIVO_ To investigate the role of NLRC5 in LPS-induced septic shock _in vivo_, we injected _NLRC5_−/−
and WT mice with high-dose _Escherichia coli_ LPS (25 mg/kg) intraperitoneally and then monitored mouse survival. Although we observed a slight, but not statistically significant,
difference in survival between the two groups after LPS treatment (Figure 7A), we found a marked increase in plasma IL-6 levels in _NLRC5_−/− mice 1 h after LPS treatment, compared with WT
mice (Figure 7B). However, such differences disappeared 3 h post LPS treatment (data not shown). We did not observe any difference in plasma TNF-α levels (data not shown). These results
provide _in vivo_ evidence that NLRC5 negatively regulates IL-6 production at early stages of LPS-induced septic shock, but not at later stages, explaining a slight, but not significant,
difference in overall mouse survival after LPS treatment. We next sought to determine whether _NLRC5_ deficiency affects antiviral responses _in vivo_. _NLRC5_−/− and WT mice were injected
with VSV-eGFP via the tail vein and then sera were collected to measure viral titers and type I IFN production. We found a significant increase in IFN-β amounts in sera of _NLRC5_−/− mice
compared to WT mice at an early time point (6 h) postinfection (Figure 7C). However, such differences gradually disappeared at 12 and 24 h postinfection (Figure 7C). Consistent with this
observation, we observed a reduction, but not at significant level, in serum virus titers in _NLRC5_−/− compared with WT group at 6, 12 and 24 h postinfection (Figure 7D). Consistently, we
found that _NLRC5_−/− mice lived a little longer than WT mice after VSV infection, but not significantly (Supplementary information, Figure S7), suggesting that _NLRC5_ deficiency increases
type I IFN production at early time point _in vivo_, and antiviral immunity but to a less extent. DISCUSSION Although NLRs were originally identified as intracellular pathogen sensors,
recent studies suggest that many NLRs can also function beyond pathogen detection10. NLRC5 has been characterized as a regulator of innate immunity and MHC class I
expression17,18,19,20,21,22,23. However, the molecular mechanisms and _in vivo_ function of NLRC5 remain to be elucidated. In particular, conflicting roles of NLRC5 in innate immune
signaling, antiviral immunity and inflammation have been reported. Thus, it is critically important to further define the function and potential mechanisms of NLRC5 in adverse biological
processes. In this study, we generated _NLRC5_-deficient mice by targeted deletion of the exon 8 of _NLRC5_, which contains the functional domain of NLRC5 that inhibits NF-κB signaling17,
while a previous report characterized mice with targeted deletion of exon 4 of _NLRC5_. Using newly generated _NLRC5_−/− mice, we provide several new lines of evidence to elucidate the
function and regulatory mechanisms of NLRC5. First, induction of _NLRC5_ expression by various TLR and cytokine stimuli is mainly controlled by Stat1-mediated pathway. _Stat1_ deletion
abolished induction of _NLRC5_ expression by stimulation with LPS or various cytokines, while NF-κB signaling pathway is required for production of several key cytokines (mainly IFN-β) that
in turn activate Stat1-mediated signaling, which ultimately control _NLRC5_ expression. Second, NLRC5 ablation markedly reduced the expression of MHC class I molecule in T cells, as well as
in B cells to a less extent, suggesting that NLRC5 plays an important role in the control of MHC class I expression. By contrast, CIITA, another member of NLR protein family, mainly controls
MHC class II expression, as well as MHC class I expression in B cells26,27. Third, our results suggest that NLRC5 regulates innate immune responses in a cell type-specific manner. NLRC5
deficiency markedly enhanced NF-κB, type I IFN signaling and antiviral immunity in MEFs, macrophages, but not in BMDCs. The cell type-specific effects of NLRC5 on innate immune signaling may
be due to the diverse expression levels of NLRC5 in different cell types as previously reported17,18,19,20, and cell type-specific signaling pathways. For example, pDCs predominantly use
the MyD88-IRF7 pathway to induce IFN-α production in response to TLR7/9 ligands and viral infection, while other cell types, including macrophages and MEFs, employ TBK1-IRF3-dependent type I
IFN signaling in response to RNA/DNA stimulation and viral infection28. Our recent studies demonstrate that TGF-β-activated kinase-1 (TAK1) is an essential positive regulator that triggers
NF-κB and MAP kinase signaling in MEFs and T cells, but exhibits the opposite effects on these signaling pathways in neutrophils29. TRAF family member-associated NF-κB activator (TANK) has
been identified as a positive regulator of transcriptional factors IRF3 and NF-κB signaling. However, a recent study showed that TANK deletion enhanced NF-κB signaling by promoting the
ubiquitination of TRAF6 upon TLR stimulation, thus serving as a negative regulator of TLR signaling30. In light of these findings, we conclude that NLRC5 functions as a negative regulator of
NF-κB and type IFN signaling in MEFs and macrophages, but has little or no effect on BMDCs. Kumar _et al._23 generated _NLRC5_ knockout mice by replacing exon 4 of _NLRC5_ with a
neomycin-resistance gene cassette, and did not observe any effect of _NLRC5_ deficiency on the induction of inflammatory cytokines and type I IFN in macrophages and dendritic cells (DCs) in
response to LPS, poly(I:C), NDV, HSV-1 and _L. monoctogenes_. Our results in BMDCs are consistent with their findings in BMDCs, showing little or no effect of _NLRC5_ deletion on innate
immune signaling in BMDCs. However, they did not observe any difference between WT and _NLRC5_−/− macrophages in response to LPS or poly(I:C) treatment. The discrepancies between our and
their studies may be due to several factors: different deletion mice of _NLRC5_ (exon 4 versus exon 8 deletion), different doses of LPS used for stimulation and different time courses of TLR
stimulation experiments. For example, Kumar _et al._ used 1 000 ng/ml LPS to stimulate macrophages, which is 10 times higher than the dose that we used (100 ng/ml). It appears that loss of
_NLRC5_ may enhance more proinflammatory cytokine release at low doses of stimulation, as large differences in IL-6 and TNF-α production were observed between _NLRC5_-deficient and WT cells
when a lower dose of LPS (10 ng/ml) was used. High doses of LPS stimulation may mask the differences in IL-6 and TNF-α production between _NLRC5_-deficient and WT cells. Moreover, since
NLRC5 inhibits NF-κB and type IFN signaling in the early time course17, we measured innate immune signaling and cytokine production at different time points. More importantly, it should be
noted that the differences between _NLRC5_-deficient and WT cells are observed only when PAMP-mediated signaling activation requires RIG-I/MDA5 and/or IKK complex. Thus, this specificity may
explain why stimulation with poly(I:C), dsDNA, HSV-1 and other pathogens showed no difference in cytokine production between _NLRC5_-deficient and WT cells. In addition, we found that NLRC5
deletion enhanced IL-6 expression in both MEFs and macrophages, but increased production of TNF-α was only observed in _NLRC5_-deficient MEFs but not in macrophages, compared with their WT
controls (Figures 3B and 5B). This might be due to multiple layers of regulation for TNF-α. It has been known that many cytokine genes such as TNF-α and IL-1β are subjected to
posttranscriptional regulation at the mRNA stability and protein translational levels31. Consistent with this, we previously showed that NLRC5 has more inhibitory effect on IL-6 than TNF-α
production in RAW264.7 cells17. Similarly, we and others recently found that NLRX1 knockdown or deletion has more dramatic effects on IL-6 than TNF-α12,13. NLRX1 knockdown results in
significantly more IL-6 production in the plasma than WT mice in response to LPS treatment, but there is no difference in TNF-α level between NLRX1 knockdown and WT mice. Although most
stimuli use the common IKK-NF-κB module to activate NF-κB signaling, the consequences of activation of downstream NF-κB-responsive genes are often different, mainly due to cell type-specific
signaling pathways, quantitative and qualitative parameters of the TLR-ligand interaction itself32. Thus, different TLR ligands may use the same NF-κB pathway, but they may produce
different amounts and/or types of cytokines in the same type of cells. Similarly, the same TLR ligand may produce different amounts and/or types of cytokines in different types of cells.
More importantly, activation and regulation of the NF-κB pathway itself may vary in a cell type-specific manner. As we recently demonstrated, TAK1 functions as a positive and essential
adaptor molecule in NF-κB and MAP kinase pathways in MEFs, but functions as a negative regulator of these pathways in neutrophils29. Thus, it is likely that MEFs use classical NF-κB pathway,
while macrophages and DCs may use the same NF-κB pathway with even increased variations. Thus, further studies are needed to address cell type-specific activation and regulation of NF-κB
and type I IFN signaling pathways. The functional redundancy of several negative regulators (such as NLRX1 and CUEDC2), which inhibit the same or similar molecules in NF-κB signaling12,33,
may compensate for the loss of NLRC5 in innate immune signaling and response to LPS and viral infection _in vivo_. Our findings show that although _NLRC5_ deficiency enhanced IL-6 and IFN-β
secretion at early time points poststimulation or infection, we observed a small, but not significant, difference in mouse survival after LPS treatment or viral infection. Recent studies
indicate that _NLRX1_−/− mice exhibited increased expression of IFN-β and IL-6 after influenza virus infection13, while another group showed no difference in type I IFN signaling between
_NLRX1_-deficient and WT mice34. It is not clear what caused these discrepancies between these studies, but it may be due to differences in cell types, duration and dose of stimuli and
different experimental system. Alternatively, generation of knockout mice harboring deletions of _NLRC5_ and _NLRX1_ may help better understand the role of these negative regulators _in
vivo_. In summary, we show that induction of _NLRC5_ expression by various stimuli including LPS and IFN-β requires Stat1-mediated signaling. _NLRC5_ ablation reduced MHC class I gene
expression in T cells and other cells, suggesting the involvement of NLRC5 in MHC class I expression. _NLRC5_ ablation enhanced NF-κB and type I IFN signaling pathways in response to LPS or
VSV infection in a cell type-specific manner. _NLRC5_-deficient mice produced higher amounts of IL-6 and IFN-β in the sera when they were challenged with LPS or infected with VSV. NLRC5
plays important roles in diverse biological processes and immune responses to pathogens in a cell type-specific manner, thus serving as important targets for modulating innate immune
signaling and regulation. MATERIALS AND METHODS GENERATION OF NLRC5-DEFICIENT MICE The targeting vector was constructed by replacing exon 8, which encodes part of the LRR1 domain of _Nlrc5_,
with a Neo cassette, leading to premature termination of NLRC5 (Figure 1A). The linearized targeting vector was injected into ES cells by electroporation. Homologous recombinant stem cells
were identified by screening with PCR and further confirmed by Southern blotting analysis. The selected homologous recombinant ES clones were injected into blastocysts. Chimeric mice
generated from two homologous ES clones were bred to generate F1 heterozygotes as well as homozygous _NLRC5_-deficient mice. All the mice were maintained in a pathogen-free animal facility,
following the Use of Laboratory Animals and the approved protocols by the IACUC committee at Baylor College of Medicine. CELL CULTURE AND TLR STIMULATION RAW264.7 cells were purchased from
ATCC (Manassas, VA) and maintained in DMEM with 10% fetal bovine serum (FBS). MEFs were prepared from day 15 embryos and cultured in DMEM supplemented with 10% FBS. _MAVS_-deficient MEFs
were previously described12. Peritoneal macrophages were harvested from mice 4 days after thioglycollate (BD, Sparks, MD) injection and were cultured in DMEM supplemented with 10% FBS. Bone
marrow cells were isolated from the tibia and femur and cultured in RPMI1640 medium with 10% FBS, 1% penicillin-streptomycin, 55 μM β-mercaptoethanol and 10% L929 conditioned media
containing macrophage-colony stimulating factor (M-CSF) for 4 days or 10 ng/ml murine GM-CSF (PeproTech) for 6-8 days to harvest BMMs or BMDCs, respectively. BMMs and BMDCs were stimulated
for the indicated times with LPS (100 ng/ml), Pam3CSK4 (1 μg/ml), CpG (2 μg/ml), CL-097 (1 μg/ml), poly(I:C) (20 μg/ml), and poly(I:C)/LyoVec (1 μg/ml), unless specifically mentioned. LPS
(_E. coli_, sterile serotype 0.111:B4) was purchased from Sigma-Aldrich. CpG ODN 1668, poly(I:C) and other TLR ligands were obtained from Invivogen (San Diego, CA, USA). FLOW CYTOMETRY
Fluorescence-conjugated antibodies against mouse CD4, CD8 and H2-kb were used in this study. All antibodies were obtained from BD Pharmingen (San Jose, CA, USA). Cells were stained, washed,
resuspended in PBS(1%) FBS(0.05%) NaN3 and analyzed using a FACSCalibur flow cytometer. All flow cytometry data were analyzed using FlowJo (TreeStar, Inc., Palo Alto, CA, USA). ANTIBODIES
Anti-IKK (sc-7607) and anti-IRF3 (sc-9082) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-β-actin was purchased from Sigma. Anti-phospho-IKK (2697),
anti-phospho-ERK (9101), anti-phospho-p38 (9211) anti-phospho-JNK (9251) and anti-phospho-IRF3 (4947) were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-NLRC5 polyclonal
antibody was previously described17. Anti-IFN-β antibody (7F-D3) was purchased from Abcam. Anti-IFN-γ antibody was obtained from clone HB170 (ATCC). IMMUNOBLOT ANALYSES For immunoblot
experiments, whole-cell extracts were lysed, boiled for 5 min with SDS loading buffer (Cell Signaling Technology, Danvers, MA, USA) and resolved on SDS-PAGE gels. The proteins were
transferred to nitrocellulose membranes (Bio-Rad), blocked with milk and further incubated with the indicated antibodies. LumiGlo Chemiluminescent Substrate System from KPL (Gaithersburg,
MD, USA) was used for protein detection. CYTOKINE RELEASE ANALYSIS MEF cells or macrophages were seeded on 24-well plates, treated with indicated stimuli and supernatants were collected and
subjected to analysis with commercial ELISA kits for mouse IL-6, TNF-α, IL-1β (eBiosceince) and IFN-β (PBL Biomedical Laboratories), following the manufacturer's instructions. For _in
vivo_ LPS septic shock experiments, mice were injected with LPS (25 mg/kg) intraperitoneally, and sera were collected before (0 h) and after (1 and 3 h) LPS injection and analyzed for their
IL-6 and TNF-α levels by ELISA. For _in vivo_ virus infection, mice were injected intravenously with VSV-eGFP. Sera were collected before (0 h) and after (3 and 6 h) viral infection and
analyzed for the IFN-β levels using ELISA. REAL-TIME PCR ANALYSIS Total RNA was harvested from MEFs or macrophages using the TRIzol reagent (Invitrogen) and the complimentary cDNA was
generated using reverse transcriptase II (Invitrogen). Real-time PCR was carried out using the ABI Prism 7000 analyzer (Applied Biosystems) using the SYBR GreenER qPCR Super Mix Universal
(Invitrogen) and specific primers. The sequences of the primers used are: Primers for mouse _Nlrc5_: forward: 5′-TCAGCCCAGAACAAGTATCC-3′; reverse: 5′-TGGGCACAGAC-TTCCATTAG-3′. Primers for
mouse _Gapdh_: forward 5′-TTGTCTCCTGCGACTTCAACAG-3′; reverse: 5′-GGTCTGGGATGGAAATTGTGAG-3′. Primers for mouse _TNF-α_: forward: 5′-ACAGAAAGCATGATCCGCG-3′; reverse: 5′-GCCCCCCATCTTTTGGG-3′.
Primers for mouse _IL-6_: forward: 5′-CCAGAAACCGCTATGAAGTTCC-3′; reverse: 5′-TTGTCACCAGCATCAGTCCC-3′. Primers for mouse _IL-1β_: forward: 5′-GTGGCTGTGGAGAAGCTGTG-3′; reverse:
5′-GAAGGTCCACGGGAAAGACAC-3′. Primers for mouse _IFN-β_: forward: 5′-TCACCTACAGGGCGGACTTC-3′; reverse: 5′-TCTCTGCTCGGACCACCATC-3′. STATISTICAL ANALYSIS Statistical significance between groups
was determined by two-tailed Student's _t_-test and two-way ANOVA test. Differences were considered to be significant when _P_ < 0.05. For mouse endotoxic shock study, Kaplan-Meier
survival curves were generated and analyzed for statistical significance with Graphpad Prism 4.0. REFERENCES * Takeuchi O, Akira S . Pattern recognition receptors and inflammation. _Cell_
2010; 140:805–820. Article CAS Google Scholar * Schroder K, Tschopp J . The inflammasomes. _Cell_ 2010; 140:821–832. Article CAS Google Scholar * Kawai T, Akira S . The role of
pattern-recognition receptors in innate immunity: update on Toll-like receptors. _Nat Immunol_ 2010; 11:373–384. Article CAS Google Scholar * Wilkins C, Gale M Jr . Recognition of viruses
by cytoplasmic sensors. _Curr Opin Immunol_ 2010; 22:41–47. Article CAS Google Scholar * Chiu YH, Macmillan JB, Chen ZJ . RNA polymerase III detects cytosolic DNA and induces type I
interferons through the RIG-I pathway. _Cell_ 2009; 138:576–591. Article CAS Google Scholar * Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V . RIG-I-dependent
sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. _Nat Immunol_ 2009; 10:1065–1072. Article CAS Google Scholar * Zhang Z, Yuan B, Bao M,
Lu N, Kim T, Liu YJ . The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. _Nat Immunol_ 2011; 12:959–965. Article CAS Google Scholar *
Unterholzner L, Keating SE, Baran M, _et al_. IFI16 is an innate immune sensor for intracellular DNA. _Nat Immunol_ 2010; 11:997–1004. Article CAS Google Scholar * Inohara N, Nunez G .
NODs: intracellular proteins involved in inflammation and apoptosis. _Nat Rev Immunol_ 2003; 3:371–382. Article CAS Google Scholar * Kufer TA, Sansonetti PJ . NLR functions beyond
pathogen recognition. _Nat Immunol_ 2011; 12:121–128. Article CAS Google Scholar * Moore CB, Bergstralh DT, Duncan JA, _et al_. NLRX1 is a regulator of mitochondrial antiviral immunity.
_Nature_ 2008; 451:573–577. Article CAS Google Scholar * Xia X, Cui J, Wang HY, _et al_. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. _Immunity_
2011; 34:843–853. Article CAS Google Scholar * Allen IC, Moore CB, Schneider M, _et al_. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS
and TRAF6-NF-kappa B signaling pathways. _Immunity_ 2011; 34:854–865. Article CAS Google Scholar * Tattoli I, Carneiro LA, Jehanno M, _et al_. NLRX1 is a mitochondrial NOD-like receptor
that amplifies NF-kappa B and JNK pathways by inducing reactive oxygen species production. _EMBO Rep_ 2008; 9:293–300. Article CAS Google Scholar * Jounai N, Kobiyama K, Shiina M, Ogata
K, Ishii KJ, Takeshita F . NLRP4 negatively regulates autophagic processes through an association with Beclin1. _J Immunol_ 2011; 186:1646–1655. Article CAS Google Scholar * Fiorentino L,
Stehlik C, Oliveira V, Ariza ME, Godzik A, Reed JC . A novel PAAD-containing protein that modulates NF-kappa B induction by cytokines tumor necrosis factor-alpha and interleukin-1 beta. _J
Biol Chem_ 2002; 277:35333–35340. Article CAS Google Scholar * Cui J, Zhu L, Xia XJ, _et al_. NLRC5 negatively regulates the NF-kappa B and type I interferon signaling pathways. _Cell_
2010; 141:483–496. Article CAS Google Scholar * Benko S, Magalhaes JG, Philpott DJ, Girardin SE . NLRC5 limits the activation of inflammatory pathways. _J Immunol_ 2010; 185:1681–1691.
Article CAS Google Scholar * Kuenzel S, Till A, Winkler M, _et al_. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune
responses. _J Immunol_ 2010; 184:1990–2000. Article CAS Google Scholar * Neerincx A, Lautz K, Menning M, _et al_. A role for the human nucleotide-binding domain, leucine-rich
repeat-containing family member NLRC5 in antiviral responses. _J Biol Chem_ 2010; 285:26223–26232. Article CAS Google Scholar * Davis BK, Roberts RA, Huang MT, _et al_. Cutting edge:
NLRC5-dependent activation of the inflammasome. _J Immunol_ 2011; 186:1333–1337. Article CAS Google Scholar * Meissner TB, Li A, Biswas A, _et al_. NLR family member NLRC5 is a
transcriptional regulator of MHC class I genes. _Proc Natl Acad Sci USA_ 2010; 107:13794–13799. Article CAS Google Scholar * Kumar H, Pandey S, Zou J, _et al_. NLRC5 deficiency does not
influence cytokine induction by virus and bacteria infections. _J Immunol_ 2011; 186:994–1000. Article CAS Google Scholar * O'Shea JJ, Gadina M, Schreiber RD . Cytokine signaling in
2002: new surprises in the Jak/Stat pathway. _Cell_ 2002; 109 SUPPL:S121–S131. Article Google Scholar * Shuai K, Liu B . Regulation of JAK-STAT signalling in the immune system. _Nat Rev
Immunol_ 2003; 3:900–911. Article CAS Google Scholar * LeibundGut-Landmann S, Waldburger JM, Krawczyk M, _et al_. Mini-review: specificity and expression of CIITA, the master regulator of
MHC class II genes. _Eur J Immunol_ 2004; 34:1513–1525. Article CAS Google Scholar * Wright KL, Ting JP . Epigenetic regulation of MHC-II and CIITA genes. _Trends Immunol_ 2006;
27:405–412. Article CAS Google Scholar * Honda K, Taniguchi T . IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. _Nat Rev Immunol_
2006; 6:644–658. Article CAS Google Scholar * Ajibade AA, Wang Q, Cui J, _et al_. TAK1 negatively regulates NF-kappaB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. _Immunity_
2012; 36:43–54. Article Google Scholar * Kawagoe T, Takeuchi O, Takabatake Y, _et al_. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of
autoimmune nephritis. _Nat Immunol_ 2009; 10:965–972. Article CAS Google Scholar * Han J, Ulevitch RJ . Limiting inflammatory responses during activation of innate immunity. _Nat Immunol_
2005; 6:1198–1205. Article CAS Google Scholar * Kanzler H, Barrat FJ, Hessel EM, Coffman RL . Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists.
_Nat Med_ 2007; 13:552–559. Article CAS Google Scholar * Li HY, Liu H, Wang CH, _et al_. Deactivation of the kinase IKK by CUEDC2 through recruitment of the phosphatase PP1. _Nat Immunol_
2008; 9:533–541. Article CAS Google Scholar * Rebsamen M, Vazquez J, Tardivel A, Guarda G, Curran J, Tschopp J . NLRX1/NOD5 deficiency does not affect MAVS signalling. _Cell Death
Differ_ 2011; 18:1387. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We would like to thank Dr Adebusola A Ajibade (The Methodist Hospital Research Institute) for
critical reading of this manuscript, and Drs Margaret A Goodell and Katherine Yudeh King (Baylor College of Medicine) for providing us with _Stat1_−/− _mice_. This work was in part supported
by Grants from National Institutes of Health (NIH) (CA090327, CA101795, CA121191, CA116408, CA094327 and DA030338), Cancer Research Institute, and The Methodist Hospital Research Institute.
Yanzheng Tong was a recipient of The China Scholarship Council (CSC). AUTHOR INFORMATION Author notes * Yanzheng Tong and Jun Cui: These two authors contributed equally to this work.
AUTHORS AND AFFILIATIONS * The Center for Cell and Gene Therapy, Baylor College of Medicine, Houston, 77030, TX, USA Yanzheng Tong, Jun Cui, Qingtian Li, Jia Zou, Helen Y Wang & Rong-Fu
Wang * Institute of Biosciences and Technology, Texas A&M University Health Science Center, Houston, 77030, TX, USA Yanzheng Tong * Center for Inflammation and Epigenetics, The Methodist
Hospital Research Institute, Houston, 77030, TX, USA Yanzheng Tong, Jun Cui, Qingtian Li, Jia Zou, Helen Y Wang & Rong-Fu Wang * Department of Pathology and Immunology, Baylor College
of Medicine, Houston, 77030, TX, USA Helen Y Wang & Rong-Fu Wang Authors * Yanzheng Tong View author publications You can also search for this author inPubMed Google Scholar * Jun Cui
View author publications You can also search for this author inPubMed Google Scholar * Qingtian Li View author publications You can also search for this author inPubMed Google Scholar * Jia
Zou View author publications You can also search for this author inPubMed Google Scholar * Helen Y Wang View author publications You can also search for this author inPubMed Google Scholar *
Rong-Fu Wang View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Rong-Fu Wang. ADDITIONAL INFORMATION ( SUPPLEMENTARY
INFORMATION is linked to the online version of the paper on the _Cell Research_ website.) SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION, FIGURE S1 Induction of _NLRC5_ expression by
the cytokines in the cell culture supernatant after LPS stimulation (PDF 633 kb) SUPPLEMENTARY INFORMATION, FIGURE S2 Southern blotting analysis of WT and _NLRC5__+/−_ embryonic stem cell
clones by (A) 5′ probe 1 and (B) 3′ probe 2 shown in Figure 2A. (PDF 41 kb) SUPPLEMENTARY INFORMATION, FIGURE S3 Enhanced TNF-α production in _NLRC5_-deficient MEF cells (PDF 264 kb)
SUPPLEMENTARY INFORMATION, FIGURE S4 _NLRC5_ deletion has little or no effect on cytokine production in BMDCs. (PDF 110 kb) SUPPLEMENTARY INFORMATION, FIGURE S5 Loss of _NLRC5_ has little or
no effect on TNF-α and IL-1β production in peritoneal macrophages and BMMs. (PDF 273 kb) SUPPLEMENTARY INFORMATION, FIGURE S6 Much more differences in IL-6 and TNF-α release between
_NLRC5_−/− and WT peritoneal macrophages cells were observed only when low, but not high, doses of LPS were used for treatment. (PDF 116 kb) SUPPLEMENTARY INFORMATION, FIGURE S7 Loss of
NLRC5 has little or no effect on mice survival or virus titer of plasma after VSV-eGFP infection _in vivo_. (PDF 46 kb) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Tong, Y., Cui, J., Li, Q. _et al._ Enhanced TLR-induced NF-κB signaling and type I interferon responses in _NLRC5_ deficient mice. _Cell Res_ 22, 822–835 (2012).
https://doi.org/10.1038/cr.2012.53 Download citation * Received: 01 February 2012 * Revised: 21 February 2012 * Accepted: 24 February 2012 * Published: 03 April 2012 * Issue Date: May 2012 *
DOI: https://doi.org/10.1038/cr.2012.53 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * innate immune signaling * NF-κB activation * type I
interferon signaling * Nod-like receptors
Trending News
Moment officer came face to face with blood-soaked killer who said 'we all die together' - YorkshireLiveNewsMoment officer came face to face with blood-soaked killer who said 'we all die together'Police Officer Jessica Witto...
The page you were looking for doesn't exist.You may have mistyped the address or the page may have moved.By proceeding, you agree to our Terms & Conditions and our ...
The page you were looking for doesn't exist.You may have mistyped the address or the page may have moved.By proceeding, you agree to our Terms & Conditions and our ...
The page you were looking for doesn't exist.You may have mistyped the address or the page may have moved.By proceeding, you agree to our Terms & Conditions and our ...
The page you were looking for doesn't exist.You may have mistyped the address or the page may have moved.By proceeding, you agree to our Terms & Conditions and our ...
Latests News
Enhanced tlr-induced nf-κb signaling and type i interferon responses in nlrc5 deficient miceABSTRACT Nod-like receptors (NLRs) are intracellular sensors that respond to a variety of pathogen and intracellular dan...
Smoking associated with work absenteeismSmokers are 33% more likely to be absent from work and take, on average, 2.7 extra sick days annually. This costs the UK...
Raman-phonon-polariton condensation in a transversely pumped cavityABSTRACT Phonon polaritons are hybrid states of light and matter that are typically realised when optically active phono...
Litigation stanches flow of data on exxon valdez oil spill's effectsANCHORAGE — A third summer has passed since the Exxon Valdez oil spill, and one can see positive signs of environmental ...
Intractable epilepsy: causes, symptoms, diagnosis, treatmentEpilepsy is a neurological disorder that causes recurring seizures. A seizure is a sudden, abnormal change in the brain’...