Toward a unified benchmark and framework for deep learning-based prediction of nuclear magnetic resonance chemical shifts
Toward a unified benchmark and framework for deep learning-based prediction of nuclear magnetic resonance chemical shifts"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
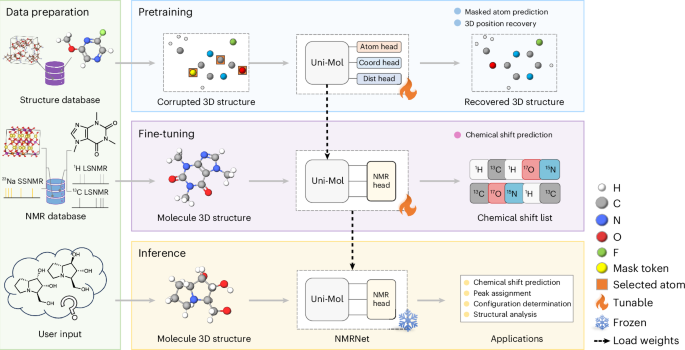
ABSTRACT The study of structure–spectrum relationships is essential for spectral interpretation, impacting structural elucidation and material design. Predicting spectra from molecular
structures is challenging due to their complex relationships. Here we introduce NMRNet, a deep learning framework using the SE(3) Transformer for atomic environment modeling, following a
pretraining and fine-tuning paradigm. To support the evaluation of nuclear magnetic resonance chemical shift prediction models, we have established a comprehensive benchmark based on
previous research and databases, covering diverse chemical systems. Applying NMRNet to these benchmark datasets, we achieve competitive performance in both liquid-state and solid-state
nuclear magnetic resonance datasets, demonstrating its robustness and practical utility in real-world scenarios. Our work helps to advance deep learning applications in analytical and
structural chemistry. Access through your institution Buy or subscribe This is a preview of subscription content, access via your institution ACCESS OPTIONS Access through your institution
Access Nature and 54 other Nature Portfolio journals Get Nature+, our best-value online-access subscription $29.99 / 30 days cancel any time Learn more Subscribe to this journal Receive 12
digital issues and online access to articles $99.00 per year only $8.25 per issue Learn more Buy this article * Purchase on SpringerLink * Instant access to full article PDF Buy now Prices
may be subject to local taxes which are calculated during checkout ADDITIONAL ACCESS OPTIONS: * Log in * Learn about institutional subscriptions * Read our FAQs * Contact customer support
SIMILAR CONTENT BEING VIEWED BY OTHERS A DEEP LEARNING MODEL FOR PREDICTING SELECTED ORGANIC MOLECULAR SPECTRA Article 13 November 2023 TRANSPEAKNET FOR SOLVENT-AWARE 2D NMR PREDICTION VIA
MULTI-TASK PRE-TRAINING AND UNSUPERVISED LEARNING Article Open access 20 February 2025 RAPID PROTEIN ASSIGNMENTS AND STRUCTURES FROM RAW NMR SPECTRA WITH THE DEEP LEARNING TECHNIQUE ARTINA
Article Open access 18 October 2022 DATA AVAILABILITY Source data for Fig. 2 and Extended Data Figs. 1, 2 and 4 are available with this Brief Communication. All structural datasets used for
pretraining are publicly accessible. The Aflow dataset32 is available at https://aflowlib.org/, the Materials Project dataset34 is accessible at https://next-gen.materialsproject.org/ and
the CSD dataset33 is accessible at https://www.ccdc.cam.ac.uk/. All processed NMR datasets used for fine tuning are available via Zenodo at https://doi.org/10.5281/zenodo.13317524 (ref. 44).
CODE AVAILABILITY The NMRNet code is available via GitHub at https://github.com/Colin-Jay/NMRNet and via Zenodo at https://doi.org/10.5281/zenodo.14741405 (ref. 45) under an open-source
license. The trained model parameters are available via Zenodo at https://doi.org/10.5281/zenodo.13317524 (ref. 44). A demo notebook of NMRNet is available at
https://bohrium.dp.tech/notebooks/38356712597, and an online service is available at https://ai4ec.ac.cn/apps/nmrnet and https://bohrium.dp.tech/apps/nmrnet001. REFERENCES * Xue, X. et al.
Advances in the application of artificial intelligence-based spectral data interpretation: a perspective. _Anal. Chem._ 95, 13733–13745 (2023). Article Google Scholar * Lu, X.-Y. et al.
Deep learning-assisted spectrum–structure correlation: state-of-the-art and perspectives. _Anal. Chem._ 96, 7959–7975 (2024). Article Google Scholar * Hu, G. & Qiu, M. Machine
learning-assisted structure annotation of natural products based on MS and NMR data. _Nat. Prod. Rep._ 40, 1735–1753 (2023). Article Google Scholar * Smith, S. G. & Goodman, J. M.
Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: the DP4 probability. _J. Am. Chem. Soc._ 132, 12946–12959 (2010). Article Google Scholar * Tsai, Y. -H. et al.
ML-_J_-DP4: an integrated quantum mechanics–machine learning approach for ultrafast NMR structural elucidation. _Org. Lett._ 24, 7487–7491 (2022). Article Google Scholar * Jonas, E.,
Kuhn, S. & Schlörer, N. Prediction of chemical shift in NMR: a review. _Magn. Reson. Chem._ 60, 1021–1031 (2022). Article Google Scholar * Cortés, I., Cuadrado, C., Hernández Daranas,
A. & Sarotti, A. M. Machine learning in computational NMR-aided structural elucidation. _Front. Nat. Prod._ 2, 1122426 (2023). Article Google Scholar * Gerrard, W. et al.
Impression–prediction of NMR parameters for 3-dimensional chemical structures using machine learning with near quantum chemical accuracy. _Chem. Sci._ 11, 508–515 (2020). Article Google
Scholar * Yang, Z., Chakraborty, M. & White, A. D. Predicting chemical shifts with graph neural networks. _Chem. Sci._ 12, 10802–10809 (2021). Article Google Scholar * Kuhn, S. &
Schlörer, N. E. Facilitating quality control for spectra assignments of small organic molecules: nmrshiftdb2—a free in-house NMR database with integrated lims for academic service
laboratories. _Magn. Reson. Chem._ 53, 582–589 (2015). Article Google Scholar * Gupta, A., Chakraborty, S. & Ramakrishnan, R. Revving up 13C NMR shielding predictions across chemical
space: benchmarks for atoms-in-molecules kernel machine learning with new data for 134 kilo molecules. _Mach. Learn. Sci. Technol._ 2, 035010 (2021). Article Google Scholar * Jonas, E.
& Kuhn, S. Rapid prediction of NMR spectral properties with quantified uncertainty. _J. Cheminform._ 11, 50 (2019). Article Google Scholar * Zou, Z. et al. A deep learning model for
predicting selected organic molecular spectra. _Nat. Comput. Sci._ 3, 957–964 (2023). Article Google Scholar * Atwi, R. et al. An automated framework for high-throughput predictions of NMR
chemical shifts within liquid solutions. _Nat. Comput. Sci._ 2, 112–122 (2022). Article Google Scholar * Paruzzo, F. M. et al. Chemical shifts in molecular solids by machine learning.
_Nat. Commun._ 9, 4501 (2018). Article Google Scholar * Lin, M. et al. Unravelling the fast alkali–ion dynamics in paramagnetic battery materials combined with NMR and deep-potential
molecular dynamics simulation. _Angew. Chem._ 133, 12655–12661 (2021). Article Google Scholar * Lin, M., Fu, R., Xiang, Y., Yang, Y. & Cheng, J. Combining NMR and molecular dynamics
simulations for revealing the alkali–ion transport in solid-state battery materials. _Curr. Opin. Electrochem._ 35, 101048 (2022). Article Google Scholar * Lin, M. et al. A machine
learning protocol for revealing ion transport mechanisms from dynamic NMR shifts in paramagnetic battery materials. _Chem. Sci._ 13, 7863–7872 (2022). Article Google Scholar * Zhou, G. et
al. Uni-Mol: a universal 3D molecular representation learning framework. In _Proc. International Conference on Learning Representations_ (eds Yan, L. et al.) (ICLR, 2023). * Kwon, Y., Lee,
D., Choi, Y.-S., Kang, M. & Kang, S. Neural message passing for nmr chemical shift prediction. _J. Chem. Inf. Model._ 60, 2024–2030 (2020). Article Google Scholar * Han, J. et al.
Scalable graph neural network for nmr chemical shift prediction. _Phys. Chem. Chem. Phys._ 24, 26870–26878 (2022). Article Google Scholar * Cordova, M. et al. A machine learning model of
chemical shifts for chemically and structurally diverse molecular solids. _J. Phys. Chem. C_ 126, 16710–16720 (2022). Article Google Scholar * Liu, S. et al. Multiresolution 3D-densenet
for chemical shift prediction in NMR crystallography. _J. Phys. Chem. Lett._ 10, 4558–4565 (2019). Article Google Scholar * Jeong, K. et al. Precisely predicting the 1H and 13C NMR
chemical shifts in new types of nerve agents and building spectra database. _Sci. Rep._ 12, 20288 (2022). Article Google Scholar * Gao, P., Zhang, J., Peng, Q., Zhang, J. & Glezakou,
V.-A. General protocol for the accurate prediction of molecular 13C/1H nmr chemical shifts via machine learning augmented DFT. _J. Chem. Inf. Model._ 60, 3746–3754 (2020). Article Google
Scholar * Wu, A. et al. Elucidating structures of complex organic compounds using a machine learning model based on the 13C NMR chemical shifts. _Precis. Chem._ 1, 57–68 (2023). Article
Google Scholar * Ai, W.-J. et al. A very deep graph convolutional network for 13C NMR chemical shift calculations with density functional theory level performance for structure assignment.
_J. Nat. Prod._ 87, 743–752 (2024). Article Google Scholar * Vergnet, J., Saubanère, M., Doublet, M.-L. & Tarascon, J.-M. The structural stability of P2-layered Na-based electrodes
during anionic redox. _Joule_ 4, 420–434 (2020). Article Google Scholar * Landrum, G. et al. Rdkit. _Zenodo_ https://doi.org/10.5281/zenodo.14779836 (2024). * Larsen, A. H. et al. The
Atomic Simulation Environment—a Python library for working with atoms. _J. Phys. Condens. Matter_ 29, 273002 (2017). Article Google Scholar * Ong, S. P. et al. Python Materials Genomics
(pymatgen): a robust, open-source Python library for materials analysis. _Comput. Mater. Sci._ 68, 314–319 (2013). Article Google Scholar * Curtarolo, S. et al. Aflow: an automatic
framework for high-throughput materials discovery. _Comput. Mater. Sci._ 58, 218–226 (2012). Article Google Scholar * Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. The
cambridge structural database. _Acta Cryst. B_ 72, 171–179 (2016). Article Google Scholar * Jain, A. et al. Commentary: the materials project: a materials genome approach to accelerating
materials innovation. _APL. Mater._ 1, 011002 (2013). Article Google Scholar * Cordova, M. et al. ShiftML. _Zenodo_ https://doi.org/10.5281/zenodo.6782653 (2022). * Luo, W. et al. Bridging
machine learning and thermodynamics for accurate p_K_a prediction. _JACS Au_ 4, 3451–3465 (2024). Article Google Scholar * Yao, L. et al. Node-aligned graph-to-graph: elevating
template-free deep learning approaches in single-step retrosynthesis. _JACS Au_ 4, 992–1003 (2024). Article Google Scholar * Abramson, J. et al. Accurate structure prediction of
biomolecular interactions with AlphaFold3. _Nature_ 630, 493–500 (2024). Article Google Scholar * Zhang, D. et al. DPA-2: a large atomic model as a multi-task learner. _NPJ Comput. Mater._
10, 293 (2024). Article Google Scholar * Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In _Proc.
NAACL-HLT_ (eds Burstein, J. et al.) (Association for Computational Linguistics, 2019). * Fang, X. et al. MolParser: end-to-end visual recognition of molecule structures in the wild.
Preprint at https://arxiv.org/abs/2411.11098v2 (2024). * Bergwerf, H. Molview: an attempt to get the cloud into chemistry classrooms. _Comm. Comput. Chem. Educ._ 9, 1–9 (2015). Google
Scholar * Momma, K. & Izumi, F. Vesta 3 for three-dimensional visualization of crystal, volumetric and morphology data. _J. Appl. Crystallogr._ 44, 1272–1276 (2011). Article Google
Scholar * Xu, F. et al. NMRNet dataset. _Zenodo_ https://doi.org/10.5281/zenodo.13317524 (2024). * Xu, F. NMRNet v1.0.0 code. _Zenodo_ https://doi.org/10.5281/zenodo.14741405 (2025).
Download references ACKNOWLEDGEMENTS We thank Y. Ren and J. Zhang for their contributions to the design of the manuscript’s cover. We thank Y. Tang and J. Qiu for his valuable improvements
to the schematic diagram. We are also grateful for the insightful discussions and suggestions from Y. Liu, J. Zou, Y. Zhuang, Y. Jin, F. Fu, W. Luo, G. Zhou and J. Wang. F.T. acknowledges
the National Key R&D Program of China (grant no. 2024YFA1210804) and a startup fund at Xiamen University. J.C. acknowledges the National Natural Science Foundation of China (grant nos.
22225302, 92470201, 22021001, 92461312, 21991151, 21991150, 92161113 and 22411560277), the Fundamental Research Funds for the Central Universities (20720220009), Laboratory of AI for
Electrochemistry (AI4EC), IKKEM (grant nos. RD2023100101 and RD2022070501). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * State Key Laboratory of Physical Chemistry of Solid Surface, College
of Chemistry and Chemical Engineering, Xiamen University, Xiamen, China Fanjie Xu, Feng Wang, Zhong-Qun Tian & Jun Cheng * DP Technology, Beijing, China Fanjie Xu, Wentao Guo, Lin Yao,
Hongshuai Wang, Zhifeng Gao & Linfeng Zhang * Department of Chemistry, University of California, Davis, CA, USA Wentao Guo * Pen-Tung Sah Institute of Micro-Nano Science and Technology,
Xiamen University, Xiamen, China Fujie Tang * Laboratory of AI for Electrochemistry, Tan Kah Kee Innovation Laboratory, Xiamen, China Fujie Tang, Zhong-Qun Tian & Jun Cheng * Institute
of Artificial Intelligence, Xiamen University, Xiamen, China Fujie Tang & Jun Cheng * AI for Science Institute, Beijing, China Linfeng Zhang & Weinan E * Center for Machine Learning
Research, Peking University, Beijing, China Weinan E * School of Mathematical Sciences, Peking University, Beijing, China Weinan E Authors * Fanjie Xu View author publications You can also
search for this author inPubMed Google Scholar * Wentao Guo View author publications You can also search for this author inPubMed Google Scholar * Feng Wang View author publications You can
also search for this author inPubMed Google Scholar * Lin Yao View author publications You can also search for this author inPubMed Google Scholar * Hongshuai Wang View author publications
You can also search for this author inPubMed Google Scholar * Fujie Tang View author publications You can also search for this author inPubMed Google Scholar * Zhifeng Gao View author
publications You can also search for this author inPubMed Google Scholar * Linfeng Zhang View author publications You can also search for this author inPubMed Google Scholar * Weinan E View
author publications You can also search for this author inPubMed Google Scholar * Zhong-Qun Tian View author publications You can also search for this author inPubMed Google Scholar * Jun
Cheng View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS J.C., F.T. and Z.G. contributed to the design of the work. F.X. and W.G. completed
data collection and cleaning. F.X. developed the NMRNet code. F.X., W.G. and Z.G. contributed to the software development. F.X., W.G. and F. T. performed data analysis. All authors
participated in the discussion and wrote the manuscript. CORRESPONDING AUTHORS Correspondence to Fujie Tang, Zhifeng Gao or Jun Cheng. ETHICS DECLARATIONS COMPETING INTERESTS The authors
declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Computational Science_ thanks Joshua D. Hartman, Nav Nidhi Rajput and the other, anonymous, reviewer(s) for their
contribution to the peer review of this work. Primary Handling Editor: Kaitlin McCardle, in collaboration with the _Nature Computational Science_ team. Peer reviewer reports are available.
ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. EXTENDED DATA EXTENDED DATA
FIG. 1 PERFORMANCE OF NMRNET IN LIQUID-STATE NMR PREDICTION. NMRNet’s correlation scatter plots of predicted versus experimental chemical shifts for (A) 1H, (B) 11B, (C) 13C, (D) 15N, (E)
17O, and (F) 19F in the nmrshiftdb2-2024 dataset. (G) Comparison of the prediction error (MAE) for different elements in the nmrshiftdb2-2024 dataset when using (marked as ‘w/ pre-training’)
or not using pre-trained (marked as ‘w/o pre-training’) weights and when predicting a single element versus all elements simultaneously. (H) Comparison of prediction error (MAE) for
different elements in the nmrshiftdb2-2024 dataset using different proportions of the training set, noting that the data volume for the 19F element does not support a 0.1% setting. To
facilitate the presentation, both the horizontal and vertical axes are scaled logarithmically. Comparison of prediction error (MAE) for different elements in (I) the nmrshiftdb2-2018 dataset
and (J) the QM9NMR dataset against previous studies. Note that DetaNet has not reported results for 19F. Source data EXTENDED DATA FIG. 2 PERFORMANCE OF NMRNET IN SOLID-STATE NMR
PREDICTION. NMRNet’s correlation scatter plots of predicted versus DFT-calculated chemical shifts (chemical shieldings) for (A) 1H, (B) 13C, (C) 15N, and (D) 17O in the ShiftML1 dataset. (E)
Distribution of chemical shifts for four elements in the ShiftML1 dataset. (F) The impact of four strategies on the prediction error (MAE) for 1H in the ShiftML1 dataset using NMRNet.
Samples represent individual atoms with labeled chemical shifts; each data point corresponds to the absolute error between predicted and actual shifts. The sample sizes (n) is 29,913. S1-S3
utilized pre-trained weights on molecular dataset, differing in their use of the unit cell with intra-cell distance matrix, the unit cell with global distance matrix, and cutoff radius = 6 Å
as the local environment for a single atom, respectively. S4 modifies the pre-training in S3 to pre-training with the cutoff format on a large-scale crystal database. (G) Comparison of the
prediction error (RMSE) for different elements in the ShiftML1 dataset using NMRNet with previous studies. (H) NMRNet’s correlation scatter plot of predicted versus DFT-calculated chemical
shifts (chemical shieldings) for 23Na in the NN-NMR dataset. Source data EXTENDED DATA FIG. 3 SIX EXAMPLES USED IN CONFIGURATION DETERMINATION. The top three are for the structure revision
task, and the bottom three are for the chiral isomer identification task. EXTENDED DATA FIG. 4 STRUCTURAL REPRESENTATIONS BY NMRNET. Local structural representations of and their
relationship with chemical shifts for all Na+ in P2-type Na2/3(Mg1/3Mn2/3)O2 using t-SNE for the (A) pre-trained NMRNet and (B) fine-tuned NMRNet. (C) Extract the interaction information
between each central atom (represented as Na1) and its local environment (Na13Mg8Mn16O39) from the results of the 64-head attention mechanism of the Transformer, each head’s results are
represented as a separate row, and these results are then concatenated together. Identical elements are arranged in ascending order based on their distances from the central atom. The darker
color in the visualization indicates stronger correlations between the central atom and its local environment. (D) A unit cell of Na2/3(Mg1/3Mn2/3)O2. (E) The local environment of Na
extracted from the infinite crystal structure corresponding to the unit cell in (D). Source data SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Notes 1 and 2, Figs. 1–14,
Tables 1–31 and additional references. PEER REVIEW FILE SOURCE DATA SOURCE DATA FIG. 2 Statistical source data for Fig. 2a,c. SOURCE DATA EXTENDED DATA FIG. 1 Statistical source data for
Extended Data Fig. 1a–j. SOURCE DATA EXTENDED DATA FIG. 2 Statistical source data for Extended Data Fig. 2a–h. SOURCE DATA EXTENDED DATA FIG. 4 Statistical source data for Extended Data Fig.
4a–c. RIGHTS AND PERMISSIONS Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or
other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Xu, F., Guo, W., Wang, F. _et al._ Toward a unified benchmark and framework for deep learning-based prediction of nuclear magnetic resonance
chemical shifts. _Nat Comput Sci_ 5, 292–300 (2025). https://doi.org/10.1038/s43588-025-00783-z Download citation * Received: 14 August 2024 * Accepted: 26 February 2025 * Published: 28
March 2025 * Issue Date: April 2025 * DOI: https://doi.org/10.1038/s43588-025-00783-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Peripheral blood regulatory T cells in patients with diffuse systemic sclerosis (SSc) before and after autologous hematopoietic SCT: a pilot studyThe present pilot study aims to evaluate the frequency and the function of regulatory T (Treg) cells in patients with di...
'The Boss' Movie Trailer - AARP2:45 AARP Videos Entertainment 'The Boss' Movie Trailer - AARP A titan of industry is sent to prison after she's caught ...
Why roger scruton is a better man than his critics | thearticleThe juggernaut of grievance which attempted to take out Sir Roger Scruton earlier in the year seems, after all, not to h...
An account of the alcyonarians collected by the royal indian marine survey ship “investigator” in the indian oceanABSTRACT THE first part of the memoir of the Alcyonarians of the Indian Ocean was published in 1906, and reviewed in NAT...
Diary of Societies | NatureARTICLE PDF RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Diary of Societies. _Na...
Latests News
Toward a unified benchmark and framework for deep learning-based prediction of nuclear magnetic resonance chemical shiftsABSTRACT The study of structure–spectrum relationships is essential for spectral interpretation, impacting structural el...
11 quick questions for actress sharon gless | members only accessNot many of her fans know that Sharon Gless almost did not become the cool, collected Christine Cagney of the 1980s prim...
Is the girl swimming 550 km for clean ganga fooling people? - scoopwhoopEleven-year-old Shraddha Shukla, who is currently undertaking the task of swimming 550 km over 10 days from Kanpur to Va...
Ankita lokhande steals a kiss from boyfriend vicky jain at a friend's wedding - video goes viralPost her bitter break-up with 'Pavitra Rishta' co-star Sushant Singh Rajput, Ankita Lokhande has found love ag...
The aarp minute: september 6, 2022Memorial Day Sale! Join AARP for just $11 per year with a 5-year membership Join now and get a FREE gift. Expires 6/4 G...