Transpeaknet for solvent-aware 2d nmr prediction via multi-task pre-training and unsupervised learning
Transpeaknet for solvent-aware 2d nmr prediction via multi-task pre-training and unsupervised learning"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Nuclear Magnetic Resonance (NMR) spectroscopy is essential for revealing molecular structure, electronic environment, and dynamics. Accurate NMR shift prediction allows researchers
to validate structures by comparing predicted and observed shifts. While Machine Learning (ML) has improved one-dimensional (1D) NMR shift prediction, predicting 2D NMR remains challenging
due to limited annotated data. To address this, we introduce an unsupervised training framework for predicting cross-peaks in 2D NMR, specifically Heteronuclear Single Quantum Coherence
(HSQC). Our approach pretrains an ML model on an annotated 1D dataset of 1H and 13C shifts, then finetunes it in an unsupervised manner using unlabeled HSQC data, which simultaneously
generates cross-peak annotations. Our model also adjusts for solvent effects. Evaluation on 479 expert-annotated HSQC spectra demonstrates our model’s superiority over traditional methods
(_ChemDraw_ and _Mestrenova_), achieving Mean Absolute Errors (MAEs) of 2.05 ppm and 0.165 ppm for 13C shifts and 1H shifts respectively. Our algorithmic annotations show a 95.21%
concordance with experts’ assignments, underscoring the approach’s potential for structural elucidation in fields like organic chemistry, pharmaceuticals, and natural products. SIMILAR
CONTENT BEING VIEWED BY OTHERS TOWARD A UNIFIED BENCHMARK AND FRAMEWORK FOR DEEP LEARNING-BASED PREDICTION OF NUCLEAR MAGNETIC RESONANCE CHEMICAL SHIFTS Article 28 March 2025 RAPID PROTEIN
ASSIGNMENTS AND STRUCTURES FROM RAW NMR SPECTRA WITH THE DEEP LEARNING TECHNIQUE ARTINA Article Open access 18 October 2022 THE 100-PROTEIN NMR SPECTRA DATASET: A RESOURCE FOR BIOMOLECULAR
NMR DATA ANALYSIS Article Open access 04 January 2024 INTRODUCTION Nuclear magnetic resonance (NMR) spectroscopy has emerged as a versatile tool with widespread applications across diverse
scientific domains, including chemistry, environmental science, food science, material science, and drug discovery by unraveling molecular dynamics and structures1,2,3,4. The primary
information of an NMR spectrum arises from the chemical shift, which is determined by the local environment of a nucleus and influenced by interactions through chemical bonds and space. This
mechanism yields unique “fingerprints” corresponding to diverse functional groups or molecular motifs, thereby facilitating the streamlined deduction of atomic connectivity and arrangement.
Interpreting NMR spectra requires following essential guidelines, often referred to as “rules of thumb”, where specific chemical shifts are associated with distinctive functional groups5.
The determination of molecular structures from varying chemical shifts on NMR spectra generally requires the expertise of experienced organic chemists. To facilitate the interpretation of
NMR spectra, significant efforts have been directed towards computational simulation of NMR spectra6. Early computational approaches, like the Hierarchically Ordered Spherical Environment
(HOSE) codes7, aim to encapsulate atom neighborhoods in concentric spheres, utilizing a nearest-neighbor approach to predict NMR shift values. A recent HOSE approach8 yields Mean Absolute
Errors (MAEs) of 3.52 ppm for 13C NMR and 0.29 ppm for 1H NMR on the nmrshiftdb29 dataset. Concurrently, significant efforts have been devoted to the ab initio calculation of NMR
properties.10,11 Density Functional Theory (DFT)-based methods were developed for certain small organic molecules, achieving MAEs of 2.9 ppm for 13C NMR and 0.23 ppm for 1H NMR12. However,
the accuracy of these DFT-based methods relies heavily on the choice of the basis functions, which often require meticulous case-by-case manual tuning for each molecule. Moreover, the
time-intensive nature of DFT calculations limits their applications to comprehensive and large datasets. Recently, the rise of Graph Neural Networks (GNN) and their successes in predicting
molecular properties13,14,15,16,17 has prompted initiatives to employ GNNs for predicting peaks in NMR spectra5,18,19. The application of GNN to molecules is intuitive, as a molecular
structure can be naturally represented as a graph, with each atom as a node and its chemical bonds as edges. On 1D NMR data, a GNN-based model achieves MAEs of 1.355 ppm for 13C NMR and
0.224 ppm for 1H NMR on the nmrshiftdb2 dataset18. While considerable efforts have been made in developing predictive models for 1D NMR, the prediction of 2D NMR remains underexplored.
Heteronuclear Single Quantum Coherence (HSQC) spectroscopy20, a sophisticated 2D NMR technique, is an important tool for elucidating atomic connectivity within complex molecules where
conventional 1D NMR may prove insufficient21,22. By correlating the chemical shifts of hydrogen nuclei with those of heteronuclear nuclei, typically carbon or nitrogen, via scalar coupling
interactions, HSQC facilitates the comprehensive mapping of interatomic connections within a molecule. This mapping yields crucial insights into chemical bonding, molecular conformation, and
intramolecular interactions. As molecular structures become more complex, 1D NMR spectra tend to display increasingly overlapping peaks, making 2D NMR techniques such as HSQC essential for
elucidating local structures. A notable stride in this domain utilizes the ML approach to establish correlations between DFT-simulated HSQC spectra and empirical data to identify
molecules23. However, the accurate prediction of HSQC spectra using ML techniques remains elusive, primarily due to the scarcity of large-scale, high-quality datasets, as well as the
labor-intensive and time-consuming peak annotation process. While numerous annotated 1D spectra are available for training ML models, combining these results to reliably generate 2D NMR data
is far from straightforward. As an example, a recent study introduced a method to integrate the state-of-the-art predictions of proton and carbon 1D spectra into HSQC spectra24, achieving
MAEs of 0.16 ppm for 1H and 2.64 ppm for 13C. This study highlights the inherent difficulties in accurately predicting 2D NMR chemical shifts, as even though the selected 1D NMR models and
methods achieve low error individually, such success cannot be transferred to HSQC cross-peak prediction. Moreover, beyond the challenge of prediction, associating each HSQC peak with its
corresponding local molecular structure is an inevitable task that requires expert domain knowledge and is often time-consuming. Proper calibration is typically necessary to ensure precise
alignment between 1D and HSQC data before interpreting the HSQC spectra. In light of the aforementioned challenges and opportunities in interpreting HSQC spectra, we propose TRANSPEAKNET: a
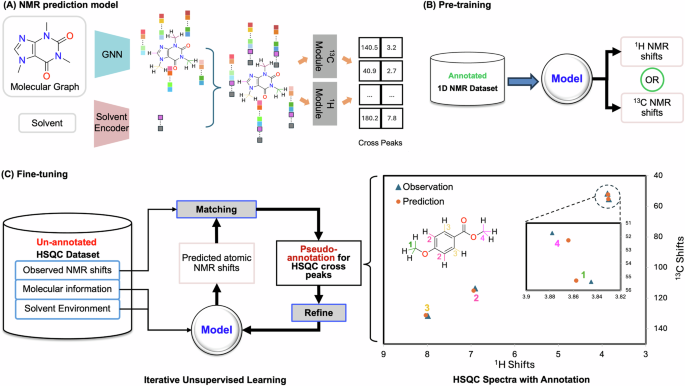
Transfer learning-based Peak prediction and assignment with unsupervised learning, illustrated in Fig. 1. This framework enables end-to-end training and testing on experimental data. Once
trained, the TransPeakNet model generates cross-peak predictions directly from a SMILES representation and the solvent environment used in the experiment. Additionally, it simultaneously
associates each cross-peak signal with its corresponding carbon-proton pairs. Alongside a Graph Neural Network (GNN) module capturing structural nuances, the model incorporates a solvent
encoder to effectively account for the impact of solvent environments on chemical shifts, which is essential for delivering accurate cross-peak prediction and peak assignment of HSQC
spectra. To tackle the lack of annotated HSQC data, we designed a two-step transfer learning process. First, the model is pre-trained on a labeled 1D NMR dataset via Multi-Task pre-Training
(MTT), enabling it to learn a wide range of C–H interactions. Then, we implement an unsupervised learning strategy that uses the unlabeled HSQC dataset to refine the model’s ability to
accurately discern and label HSQC cross peaks. The model is pre-trained using ~24,000 annotated 1D NMR dataset from NMRShiftDB29, and finetuned on ~19,000 experimental HSQC spectra from
HMDB25 and CH-NMR-NP26. The model is thoroughly evaluated and compared to traditional tools like _ChemDraw_27 and _Mestrenova_28 using expert-annotated test datasets. On test dataset, the
model achieves MAEs of 2.05 ppm and 0.165 ppm for 13C shifts and 1H shifts respectively. We also demonstrate that our model effectively considers the impact solvent has on chemical shift,
when making the prediction. When compared with the traditional tools, our model shows promising improvements, especially as the molecular size becomes larger. RESULTS PERFORMANCE ON HSQC
CROSS PEAK PREDICTION AND ASSIGNMENT Figure 2 summarizes the performance of our model on the tasks of HSQC cross-peak prediction and peak assignment, using an expert-annotated test dataset
consisting of 500 molecules, with an average molecular weight of 398.98 Da., and an average number of 56.32 atoms. The annotation process involved three experienced experts with extensive
knowledge in organic chemistry. For each molecule, two experts independently linked the observed cross-peaks from experiments to C–H bonds. If they agreed, the annotation was finalized. In
cases of disagreement, the third expert reviewed and validated the annotations. Samples with poor quality, such as those with insufficient experimental resolution, were excluded from the
test dataset for model evaluation, resulting in 479 high-quality annotations. For chemical shift prediction, our model achieved an MAEs of 2.05 ppm for 13C shifts and 0.16 ppm for 1H shifts.
In terms of annotation accuracy, our model accurately annotated all peaks in 456 out of 479 molecules (95.21%). An example of the model prediction and annotation of a small molecule is
visualized in Fig. 2C for simplicity. Additional examples for medium and large molecules are included in Supplementary Information Section 1, Supplementary Figs. 1–4. For those 23 molecules
that our algorithmic annotations do not fully agree with the experts, 81.56% of the peak annotations still align. Examples of partially correct annotations are visualized in Supplementary
Information Section 2, Supplementary Figs. 5–9. Notably, the peak assignment algorithm operates without a shift discrepancy threshold, allowing it to align all ground truth peaks with
predictions. Given the low MAE of the predicted shifts (2.05 ppm for 13C and 0.165 ppm for 1H), the model provides a solid foundation for accurate assignment. Even in rare cases where a few
atoms in a molecule have relatively large prediction errors, the annotation remains reliable, highlighting its robustness. To evaluate our model’s ability to capture solvent effects, we
tested different solvent encoders for carbon and hydrogen atoms. Empirical results indicate that a solvent encoder with a dimension of 32 is most effective at capturing proton shifts, while
adding additional embeddings for carbon does not yield significant benefits. This partially aligns with expert expectations, as protons are known to be more sensitive to their solvent
environment due to their high exposure to the surrounding molecular environment. By contrast, carbon atoms are less directly influenced by solvent interactions, as they are more deeply
embedded within the molecular structure and shielded by surrounding electron clouds. Additionally, carbon atoms are not directly involved in hydrogen bonding, and their larger mass and lower
sensitivity to external magnetic fields make their shifts less responsive to subtle solvent changes. Furthermore, since the 1D NMR data used for pre-training does not include solvent
information, and some solvent groups (e.g., benzene, acid, etc.) in the HSQC data are underrepresented, as shown in Fig. 3A, the influence of solvent on carbon shifts may be too subtle to
capture at this stage. This remains as our ongoing area of investigation for further insights. To assess the effect of solvents on proton shifts, we report model performance using the true
experimental solvent, a random solvent condition, and the “unknown” solvent condition, with the comparison shown in Fig. 3B. The results demonstrate that using the correct solvent input
yields the lowest prediction error, aligning most closely with experimental observations. This improvement in prediction is particularly significant for the CCl3, DMSO, and methanol solvent
classes, likely due to their prominent presence in the dataset. The effect of water, on the other hand, despite constituting only 1.02% of the data, can be effectively captured by the
solvent encoder. This could be explained by water’s similarity to methanol in its behavior as a hydrogen-bond donor or acceptor. Both solvents can strongly deshield protons in solutes,
causing their NMR peaks to shift higher. The predictability of these hydrogen-bonding interactions may enable the model to generalize well, even with relatively few training examples for
water. These findings underscore the promise of incorporating solvent environments into peak prediction models and highlight the need for more high-quality data with solvent information.
COMPARISON WITH TRADITIONAL TOOLS In organic chemistry, simulating HSQC spectra is crucial for analyzing experimental HSQC spectra, as it assists researchers in assigning the observed cross
peaks to the C–H bonds in target molecules. Traditional approaches, including software solutions such as _ChemDraw_27, and _Mestrenova_28, have long served as the primary resources for this
task. We compared our model with _ChemDraw_ and _Mestrenova_ and the results are shown in Fig. 4, which clearly demonstrate the superiority of our model. It is labor intensive to align
predictions from these traditional tools with ground truth. Moreover, these tools could yield unstable predictions as molecular size and complexity increases. Hence, we randomly selected 150
peaks across different molecular weight categories for comparison. Because the HSQC prediction is not inherently implemented in _ChemDraw_, the cross peaks were constructed and assigned
using 1D predictions (13C and 1H chemical shifts) based on bond connectivity. We also provided two examples with different molecular sizes to visualize the comparison. PERFORMANCE BY
SEGMENTATION To comprehensively evaluate the robustness and generalizability of our model, we conducted a segmentation analysis in the test dataset, assessing its performance across various
molecular subcategories. Segmentation is crucial in this context because it allows us to determine whether the model performs consistently well across diverse molecular characteristics,
rather than excelling in only a specific subset. We selected categories based on molecular weight and the presence of saccharides. These categories were chosen because NMR prediction gets
increasingly challenging as the molecule gets larger, while saccharides represent a distinct chemical group with unique structural features. By examining the model’s performance within these
defined segments, we aim to demonstrate its universal applicability and robustness across a wide range of molecular types. Molecular Weight (MW) is a general indicator of a molecule’s
complexity, encompassing varied geometries, bonding patterns, and the presence of isomers. These factors contribute to increased intramolecular interactions, resulting in spectral complexity
such as closely spaced peaks or overlapping signals. Additionally, the increased number of spin-spin interactions within larger molecules necessitates more advanced NMR techniques to
achieve sufficient resolution29. Furthermore, solubility issues can lead to weak signals, further complicating spectral analysis. Consequently, interpreting HSQC spectra for medium and large
molecules is challenging. Therefore, there is a pressing need for a model that can effectively predict and analyze HSQC spectra for these complex molecules. Figure 5A showcases the
stratified performance on this category, where the test molecules are grouped into three categories: small (MW < 500 daltons), medium (500 <= MW < 1000 daltons), and large (1000
daltons <= MW). On the task of predicting 1H shifts of HSQC cross peaks, the model performs comparably across all groups, achieving excellent MAE of 0.16–0.19 ppm. On the task of
predicting 13C shifts, the model achieves an MAE of 1.93 ppm for medium-sized molecules. Our model demonstrates a good generalization power on large molecules and achieves an MAE of 2 ppm on
predicting 13C shifts for this category, despite the training data containing only a small proportion of large molecules (~2%). Saccharides, or carbohydrates, play critical roles in various
biological processes involved in energy source and storage, cell signaling, cell adhesion, cell recognition, structural integrity of cells and tissues, as well as cognitive functions and
metabolic regulation30,31,32,33. Despite their importance, elucidating the structures of saccharides is challenging due to their inherent structural complexity and diversity. This complexity
arises from the diverse arrangements of monosaccharide units, varied anomeric configurations, and variable glycosidic linkages. Additionally, carbohydrates often lack the crystallinity
required for high-resolution X-ray diffraction, unlike the well-defined crystalline structures of small molecules or proteins. Consequently, NMR spectroscopy, particularly through techniques
such as HSQC, has emerged as an indispensable tool in unraveling the detailed structures of carbohydrates34,35. Forecasting HSQC cross peaks and aligning them with experimental data can
assist in comprehending saccharide connectivity and stereochemistry, thus aiding in structural determination. Our model demonstrates excellent performance in predicting HSQC cross peaks for
saccharides molecules, yielding MAEs of 1.78 for 13C shifts and 0.16 for 1H shifts (see Fig. 5(B)). This level of accuracy is consistent with the overall model performance, which
demonstrates the model’s robustness in handling complex saccharide structures. Figure 6 shows the performance of our model on a few exemplar saccharides. These saccharides feature multiple
ring structures and numerous stereogenic centers, contributing to the intricate nature of their HSQC spectra. Despite these inherent complexities, our model exhibits high accuracy in
predicting the HSQC cross peaks for these molecules. This robust performance underscores our model’s capacity to navigate the complexities associated with saccharides, thereby emphasizing
its versatility and effectiveness across various applications in the field. EFFECTS OF PRE-TRAINING AND FINE-TUNING After pre-trained via MTT on the 1D NMR dataset, the model achieved the
validation performance with MAEs of 0.210 ppm for 1H NMR prediction and 2.228 ppm for 13C NMR prediction. This success can be attributed to MTT which allows the model to effectively learn
atomic latent features as well as local structural information by simultaneously performing 1H and 13C NMR shift predictions. This helps us surpass the problem with limited annotated HSQC
data. However, when directly deploying the pre-trained model on HSQC test dataset, the model MAEs increase to 1.397 ppm and 2.822 ppm for 1H and 13C shifts, respectively. These relatively
large MAEs are expected as the data distribution of the HSQC dataset (76.34% small molecules and 90.33% non-saccharides) differs significantly from that of the 1D NMR dataset (98.80% small
molecules and 99.95% non-saccharides). In addition, the HSQC cross peaks involve interactions beyond simple pairings of 1D 13C and 1H shifts, requiring a deeper understanding of interactions
between atoms. Finally, the frequent absence of solvent labels in the 1D NMR dataset prevents the model from learning solvent effects. Nevertheless, the pre-training via MTT offers a robust
foundation for fine-tuning the model via unsupervised transfer learning. With each iteration, we observed a reduction in model errors. The performance improvement is more pronounced during
the initial iterations and gradually diminishes. By the fifth iteration, the improvement became marginal, indicating the convergence of fine-tuning. Finally, the fine-tuned model achieves
MAEs of 0.165 ppm and 2.05 ppm for 1H and 13C shifts, respectively. Throughout the transfer learning process, the model was trained to gain a more profound understanding of solvent effects
and complex C–H interactions due to intricate molecular structures. DISCUSSION In this study, we introduce a framework to develop machine learning techniques for predicting C–H cross peaks
in HSQC spectra, The framework enables us to tackle two major challenges in this avenue. The first challenge is the scarcity of annotated HSQC data for training machine learning models. The
second challenge is that collecting large volumes of annotated HSQC data is labor-intensive and requires highly trained personnel. In implementing our framework, we developed a model
combining a GNN with a solvent encoder. The GNN is trained to generate atomic embeddings that encapsulate both the local and global chemical environments of each atom, which is crucial for
accurate chemical shift predictions. The atomic embeddings are combined with the solvent embedding produced by the solvent encode, which allows our model to learn the influence of solvent on
chemical shifts. The combined embeddings are mapped by the Multi-Layer Perceptron (MLP) modules to HSQC chemical shifts. Our framework employs a two-stage transductive strategy to train the
model while addressing the aforementioned challenges. In the first stage, we use a large amount of annotated 1D NMR data to pre-train the model via Multi-Task learning. This enables the
model to adeptly grasp the intricate relationship between atomic interactions and NMR signals, laying a robust foundation for the subsequent stage. Next, the model is refined on a set of
unlabeled HSQC spectra via Iterative Unsupervised Learning, enhancing the model’s capability in predicting and interpreting HSQC spectra. Our final model achieves MAEs of 0.165 ppm and 2.05
ppm for 1H and 13C shifts respectively, while accurately assigning cross peaks. It demonstrates a consistent performance across various molecular weight and saccharide categories,
significantly outperforming the traditional methods, and shows convincing generalization capabilities to less represented samples from the training dataset. In the future, we plan to refine
our model by developing 3D-GNN models that are able to consider 3D structural information such as spatial orientation and conformational flexibility. This enhancement should enable us to
handle other 2D NMR spectra, such as Correlation Spectroscopy and Nuclear Overhauser Effect Spectroscopy, thus broadening its applicability and making a more substantial contribution to the
field of chemical analysis. METHODS In this section, we explain the components of our model and the training strategy in detail. DATA The pre-training dataset used in the MTT process is a 1D
NMR dataset from NMRShiftDB29, which contains ~24,000 annotated NMR spectra collected from 22,663 distinct molecules. The datasets used in the unsupervised transfer learning process
consist of a training dataset containing ~19,000 experimental HSQC spectra and a validation dataset containing ~5000 HSQC spectra, collected from HMDB25 and CH-NMR-NP26. All data was
accessed in September 2023, with no evidence of potential bias. To prevent data leakage in the validation dataset, duplicated spectra and molecules were removed. RDKit package in Python is
used to perform sanity check for all SMILES strings to generate valid molecular topology graph. To quantitatively evaluate our model, we built a test dataset by randomly selecting 500
spectra and manually annotating them to establish the ground truth (see Section “Performance on HSQC cross peak prediction and assignment” for the annotation process). Additionally, to
compare our model with two conventional tools (ChenDraw and Mestrenova) in chemistry, we randomly selected several molecules from this test dataset, consisting ~150 cross-peaks. Since it is
labor-intensive to derive HSQC shifts from molecular formulas using these established tools, stratified sampling was used to select these samples, ensuring the coverage of different
molecular weight groups (0–499 Dalton, 500–999 Dalton, and 1000+ Dalton). The comparison results are presented in Section “Comparison with traditional tools”. 2D NMR PREDICTION MODEL As
illustrated in Fig. 1A, our model contains a GNN component for encoding molecular features and a solvent encoder component for embedding solvent information. The GNN component learns atomic
embeddings that capture both the local and global chemical environments of each atom, which are essential for understanding the observed NMR chemical shifts. The learnt atom representations
are expanded by the solvent embedding, and then are mapped to 13C and 1H cross peaks by a MLP component. GNN A molecule can be represented by a graph _G_ = (_V_, _E_), where _V_ is the node
set representing atoms and _E_ is the edge set representing chemical bonds. Three features are provided for each node: atomic type, chirality, and hybridization. Also, two features are
considered for each edge: bond type and bond direction. Bond types include Single, Double, Triple, and Aromatic, each reflecting a distinct configuration of electron sharing between atoms.
Bond direction includes None, EndUpRight, and EndDownRight, primarily representing stereochemistry in double bonds. Each atom’s feature vector is embedded into a representation vector by a
learnable encoder. Similarly, each edge’s feature vector is embedded into a representation vector of the same length by another learnable encoder. Then, a GNN model36,37,38,39,40 utilizes
the message passing mechanism to iteratively refine the representation of each node based on information from its neighbors and connected edges. This mechanism allows the learnt node
representation to effectively capture structural context, reflecting the foundational principles of atomic interactions. Our implementation of the message passing mechanism is illustrated in
Fig. 7. It iterates for a predefined number of layers _L_, facilitating the propagation of information throughout the graph. Consequently, each node can gradually accumulate information
from a wider neighborhood across successive layers. This allow the final representation of each node to capture both local and global structural information. Our model features 5 GNN layers,
with an atomic embedding dimension of 512. SOLVENT ENCODER Since the solvent has a profound impact on NMR chemical shifts, we incorporated a trainable solvent encoder component into our
model to accurately capture this influence. We identified the following 9 principal solvent groups based on their prevalence in our dataset and domain-specific understandings of their
distinct impacts on NMR shifts. These groups include trichloromethane, dimethyl sulfoxide, acetone, acids, benzene, methanol, pyridine, water, and an additional category to encompass any
unspecified solvents from our dataset (termed “unknown”). The solvent encoder transforms each discrete solvent group _i_ into a unique, dense feature vector \({S}_{i}^{d}\), where _d_ is the
embedding dimension. These learnable vectors are optimized alongside other model parameters during training, resulting in representations that accurately reflect the impact of each solvent
class. Given the different sensitivities of carbon (C) and hydrogen (H) nuclei to solvent environments, different embedding dimensions _d_ can be chosen to tailor the solvent effect modeling
for each nuclei type. A larger embedding dimension _d_ allows the embedding to more effectively capturing the nuanced influence of solvents on NMR shifts. In our implementation, the solvent
embedding dimension for hydrogen (H) is set to 32. ATOMIC NMR SHIFT PREDICTION Finally, the embedding of each atom \({h}_{v}^{(L)}\) and the solvent embedding \({S}_{i}^{d}\) for each
solvent class _i_ are concatenated to produce a holistic representation of the atom within the context of its molecular structure and the given solvent. This combined representation is
subsequently processed by a MLP network to predict the NMR shifts for the atom: $${y}_{v}=\,{\mbox{MLP}}\,({{{\bf{h}}}}_{v}^{(L)}\oplus {{{\bf{S}}}}_{i}^{d})$$ (1) where _y__v_ is the
predicted chemical shift of atom _v_, \({h}_{v}^{(L)}\) is the atom level embedding produced by GNN, _S__d_ is the solvent embedding, and ⊕ is the concatenation operation. By integrating
solvent embedding and atomic embedding, the model effectively combines intrinsic molecular properties and solvent effects, enhancing its ability to predict atomic NMR shifts accurately. Two
separate MLP modules are used for predicting 13C and 1H shift in the cross peak predictions, respectively. Each C atom can bond up to 4 H atoms. When bonded to one, three, or four H atoms, a
C atom typically shows only one cross peak in an experimental spectrum. However, when a C atom is connected to two H atoms, up to two cross peaks may be observed, depending on the chiral
center. Consequently, a C atom can exhibit at most two 13C and 1H cross peaks. In light of this observation, one MLP module is dedicated to predicting the 13C shifts and another MLP module
for the corresponding 1H shifts. For cross peak predictions, the 13C shifts are predicted using the embeddings of C atoms. The corresponding 1H shift predictions for each C atom incorporate
aggregates of embeddings from all bonded H atoms, resulting in two predictions that are typically very similar when only one cross peak is theoretically possible. This design enhances the
model’s accuracy in predicting 1H shifts by leveraging the C atom-centered aggregation of the H atom context. By integrating the contextual dynamics around each C atom, the model provides a
more detailed and accurate mapping of hydrogen environments, crucial for pinpointing precise cross peaks in complex HSQC spectra. In our implementation, we used 2 MLP layers, with the hidden
dimensions to be 128 and 64 respectively. The dropout mechanism, which randomly deactivates a subset of neurons during the forward pass, is employed during training to prevent overfitting.
By reducing the model’s dependence on specific neurons, dropout encourages the model to learn more robust and generalized patterns. During inference, dropout is typically disabled to ensure
deterministic predictions. However, when enabled during inference, dropout can be leveraged to estimate uncertainty by calculating the standard deviation across multiple predictions18. An
example of this uncertainty estimation is provided in Supplementary Information, Section 4, Supplementary Table 1. TRAINING STRATEGY The cross peaks are notably sparse in an HSQC spectrum,
where typical resolutions for 13C and 1H shifts are 0.1 and 0.01 ppm, respectively. A typical HSQC spectrum can include 20,000 readings, covering 13C shifts from 0 to 200 ppm and 1H shifts
from 0 to 10 ppm. However, almost all of these readings are zeros, with only a small fraction representing the potential cross peaks of C–H bonds, crucial for molecular structure analysis.
Moreover, the scarcity of annotated HSQC data, particularly the labor-intensive annotations that link cross peaks to C–H bonds, makes model training difficult. To deal with this issue, we
deployed MTT to pre-train the model using an extensive annotated 1D NMR dataset (Fig. 1B). This step acclimates the model with a broad range of molecular structures and their chemical
shifts, and enables it to capture the intricate interplay between molecular structures and their NMR characteristics. Subsequently, we utilize an unsupervised strategy to refine the model
iteratively on the HSQC dataset (Fig. 1C). Through iterative cycles of prediction, annotation, and re-training, the model progressively enhances its understanding of the complex
relationships and patterns within the HSQC spectra, thus improving its predictive accuracy and providing precise cross peak alignments. By combining the MTT and unsupervised transfer
learning, we extend our annotation capabilities from 1D to 2D data, thereby enhancing the model’s predictive power and utility as a robust tool for NMR spectra analysis. PRE-TRAINING ON 1D
NMR DATA In the pre-training phase, we utilized approximately 24,000 annotated 1D NMR data points. Among these, around 22,000 samples exclusively feature 13C shifts, approximately 400
samples solely exhibit 1H shifts, while roughly 1600 samples contain both 1H and 13C shifts. To train the model effectively for predicting both 1H and 13C shifts, we adapt the MTT approach,
which enables simultaneous training on multiple related tasks. When the input data contains 13C shifts, the model predicts only carbon shifts and assesses the errors between the predicted
and actual values. Conversely, when the data sample contains 1H shifts, the hydrogen shift prediction module is activated. In both scenarios, the embeddings of 13C and 1H atoms in the GNN
module are updated simultaneously, benefiting from the message passing mechanism. Therefore, the learnt representations implicitly contain a basic understanding of C–H relationships,
essential for the interpretation of HSQC data. However, the relative scarcity of 1H shift data, due to the difficulties in accurately obtaining and extracting peaks 1H from experimental
data, complicates the training process as focusing extensively on one type of shift could compromise the model’s ability to accurately predict the other. To handle this problem in the MTT
training, we performed over-sampling on a subset of data that contain both 1H and 13C shifts, and those containing only 1H shifts. Consequently, the learned representations develop a
fundamental understanding of C–H relationships, crucial for interpreting HSQC data effectively. This integration of learned atomic relationships streamlines the transition to HSQC cross peak
predictions, thereby enhancing the model’s accuracy and efficiency in analyzing HSQC spectra. UNSUPERVISED FINE-TUNING ON HSQC DATA The model pre-trained on the 1D NMR dataset has limited
ability to predict HSQC cross-peaks from molecular structures due to the differences in data pre-processing and data distribution. First, in the 1D NMR data from NMRShiftDB2, the chemical
shifts of non-singlet peaks are averaged as ground truth. For example, whether the group is methine (-CH), methylene (-CH2), or methyl (-CH3), the proton shifts may be averaged into a single
value. In contrast, HSQC data captures C-H bonds and typically displays two cross-peaks for prochiral methylene groups due to the different environments of the two hydrogens. Additionally,
the proton chemical shifts of HSQC or HMQC cross peaks represent 13C-bound protons, whereas the signals in the 1H mainly represent 12C-bound protons, potentially leading to subtle
differences in chemical shift values41. Second, the molecule distributions in our 1D NMR data and HSQC data exhibit significant differences (see Supplementary Information, Section 3,
Supplementary Fig. 10, for data distribution plots). The HSQC dataset comprises 76.34% small molecules and 90.33% non-saccharides, whereas the 1D NMR dataset contains 98.80% small molecules
and 99.95% non-saccharides. Lastly, solvent information is not available in 1D NMR dataset, and is recorded as “unknown” in the modeling framework, whereas most molecules in the HSQC dataset
are associated with known solvent environments. This makes the fine-tuning step essential for the success of our solvent-aware framework. However, the HSQC dataset is not annotated. In
response, we implement an unsupervised training strategy (Fig. 1C), which iterates between (a) aligning cross peak prediction from the model with the experiment observations to annotate the
HSQC data and (b) using the newly acquired annotations to fine-tune the NMR prediction model, until convergence. PSEUDO-ANNOTATION OF HSQC At the end of each round in the unsupervised
learning process, the model’s predicted signals are aligned with the experimental observations to create pseudo-labels. In straightforward cases where the number of C–H bonds in a molecular
graph matches the observed HSQC cross peaks, the Hungarian algorithm42,43 is used. This classic optimization technique solves assignment problems by minimizing the cost of matching a set of
predictions to a set of observations. In the context of NMR analysis, the “cost” is defined as the discrepancy between the predicted chemical shifts and the actual shifts observed
experimentally. By systematically reducing these differences, the Hungarian algorithm achieves an optimal one-to-one correspondence between predicted shift pairs and experimental signals,
even in complex scenarios with potential signal overlap. However, in most cases, the number of C–H bonds within a molecule exceeds the number of signals recorded, making peak alignment more
difficult. This mismatch in numbers arises from several factors: firstly, rotational equivalence can reduce the number of signals, with a single peak representing all three C–H bonds for
methyl groups; secondly, symmetrical molecular structures can result in a single detectable signal for multiple symmetric C–H bonds, as seen in benzene molecule where only one peak
represents all six C–H bonds; lastly, in highly complex molecules, overlapping signals obscure some peaks, reducing the detectability of individual C–H bonds from experiments. To overcome
this issue, we utilize the graduated assignment algorithm16,44, which facilitates matching between graphs of different node counts, making it particularly suitable for this scenario. In this
algorithm, our model’s predicted C–H shifts \({({C}^{i},{H}^{i})}_{i = 0}^{N}\) and the observed C–H signals \({({C}^{j},{H}^{j})}_{j = 0}^{M}\) of each molecule are conceptualized as
points on a 2D plane, where _N_ and _M_ are numbers of predicted and observed C–H shifts respectively. These points are then treated as vertices in two fully connected graphs, _G_1 for
predicted shifts and _G_2 for observed signals. The similarity between nodes is defined as the inverse of differences between predicted chemical shifts (node in _G_1) and observed shifts
(node in _G_2). Specifically, for each predicted shift, we compute its difference with every observed shift, where a smaller difference indicates a higher similarity. The derive the
assignment matrix _A_ where each element _A__u__v_ ∈ {0, 1} indicates whether node _u_ in _G_1 matches with node _v_ in _G_2, the algorithm first finds the soft matching matrix that relaxes
the binary constraint _A__u__v_ ∈ {0, 1} to a continuous range [0, 1], then converts it into hard assignment in a greedy way, enabling one-to-many matching. DATA AVAILABILITY HSQC data used
for model performance testing is available here(via Google Drive), and the training and validation datasets will be available upon request. CODE AVAILABILITY The code is available in Github:
https://github.com/siriusxiao62/2dNMR.git REFERENCES * Gunther, H. & Gunther, H. _NMR spectroscopy: basic principles, concepts, and applications in chemistry_ (John Wiley & Sons
Chichester, UK, 1994). * Claridge, T. D. _High-resolution NMR techniques in organic chemistry_, vol. 27 (Elsevier, 2016). * Yu, H.-Y., Myoung, S. & Ahn, S. Recent applications of
benchtop nuclear magnetic resonance spectroscopy. _Magnetochemistry_ 7, 121 (2021). Article CAS Google Scholar * Lin, E. et al. High-resolution reconstruction for multidimensional laplace
NMR. _J. Phys. Chem. Lett._ 12, 5085–5090 (2021). Article CAS PubMed PubMed Central Google Scholar * Jonas, E. & Kuhn, S. Rapid prediction of nmr spectral properties with
quantified uncertainty. _J. Cheminf._ 11, 1–7 (2019). Article CAS Google Scholar * Jonas, E., Kuhn, S. & Schlörer, N. Prediction of chemical shift in NMR: A review. _Magn. Reson.
Chem._ 60, 1021–1031 (2022). Article CAS PubMed Google Scholar * Bremser, W. Hose-a novel substructure code. _Analytica Chim. Acta_ 103, 355–365 (1978). Article CAS Google Scholar *
Kuhn, S. & Johnson, S. R. Stereo-aware extension of hose codes. _ACS Omega_ 4, 7323–7329 (2019). Article CAS PubMed PubMed Central Google Scholar * Steinbeck, C., Krause, S. &
Kuhn, S. NMRShiftDB constructing a free chemical information system with open-source components. _J. Chem. Inf. Comput. Sci._ 43, 1733–1739 (2003). Article CAS PubMed Google Scholar *
Lodewyk, M. W., Siebert, M. R. & Tantillo, D. J. Computational prediction of 1h and 13c chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry.
_Chem. Rev._ 112, 1839–1862 (2012). Article CAS PubMed Google Scholar * Willoughby, P. H., Jansma, M. J. & Hoye, T. R. A guide to small-molecule structure assignment through
computation of (1h and 13c) nmr chemical shifts. _Nat. Protoc._ 9, 643–660 (2014). Article CAS PubMed Google Scholar * Wiitala, K. W., Hoye, T. R. & Cramer, C. J. Hybrid density
functional methods empirically optimized for the computation of 13c and 1h chemical shifts in chloroform solution. _J. Chem. Theory Comput._ 2, 1085–1092 (2006). Article CAS PubMed Google
Scholar * Wieder, O. et al. A compact review of molecular property prediction with graph neural networks. _Drug Discov. Today.: Technol._ 37, 1–12 (2020). Article PubMed Google Scholar
* Zhang, Z. et al. Graph neural network approaches for drug-target interactions. _Curr. Opin. Struct. Biol._ 73, 102327 (2022). Article CAS PubMed Google Scholar * Fang, X. et al.
Geometry-enhanced molecular representation learning for property prediction. _Nat. Mach. Intell._ 4, 127–134 (2022). Article Google Scholar * Wang, Y. et al. Motif-based graph
representation learning with application to chemical molecules. In _Informatics_, vol. 10, 8 (MDPI, 2023). * Xu, H., Zhou, Z. & Hong, P. Graph multi-similarity learning for molecular
property prediction. _ICML 2024 AI for Science Workshop_ (2024). * Kwon, Y., Lee, D., Choi, Y.-S., Kang, M. & Kang, S. Neural message passing for nmr chemical shift prediction. _J. Chem.
Inf. Model._ 60, 2024–2030 (2020). Article CAS PubMed Google Scholar * Guan, Y., Sowndarya, S. S., Gallegos, L. C., John, P. C. S. & Paton, R. S. Real-time prediction of 1 h and 13
c chemical shifts with dft accuracy using a 3d graph neural network. _Chem. Sci._ 12, 12012–12026 (2021). Article CAS PubMed PubMed Central Google Scholar * Bodenhausen, G. & Ruben,
D. J. Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. _Chem. Phys. Lett._ 69, 185–189 (1980). Article CAS Google Scholar * Bross-Walch, N., Kühn, T., Moskau, D.
& Zerbe, O. Strategies and tools for structure determination of natural products using modern methods of NMR spectroscopy. _Chem. Biodivers._ 2, 147–177 (2005). Article PubMed Google
Scholar * Li, Q. & Kang, C. A practical perspective on the roles of solution nmr spectroscopy in drug discovery. _Molecules_ 25, 2974 (2020). Article CAS PubMed PubMed Central
Google Scholar * Zanardi, M. M. & Sarotti, A. M. GIAO C–H COSY cosy simulations merged with artificial neural networks pattern recognition analysis. pushing the structural validation a
step forward. _J. Org. Chem._ 80, 9371–9378 (2015). Article CAS PubMed Google Scholar * Priessner, M. et al. HSQC spectra simulation and matching for molecular identification. _J. Chem.
Inf. Model._ 64, 3180–3191 (2024). Article CAS PubMed Google Scholar * Wishart, D. S. et al. HMDB 5.0: the human metabolome database for 2022. _Nucleic Acids Res._ 50, D622–D631 (2022).
Article CAS PubMed Google Scholar * Asakura, K. A NMR spectral database of natural products CH-NMR-NP. _J. Synth. Org. Chem. Jpn._ 73, 1247–1252 (2015). * Mills, N. ChemDraw Ultra 10.0
CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. _Chem. Eng. News_ 84, 13649–13650 (2006). * Willcott, M. R. MestReNova. _J. Am. Chem. Soc._ 131, 13180 (2009). * Foster, M. P.,
McElroy, C. A. & Amero, C. D. Solution nmr of large molecules and assemblies. _Biochemistry_ 46, 331–340 (2007). Article CAS PubMed Google Scholar * Guillén, D., Sánchez, S. &
Rodríguez-Sanoja, R. Carbohydrate-binding domains: multiplicity of biological roles. _Appl. Microbiol. Biotechnol._ 85, 1241–1249 (2010). Article PubMed Google Scholar * Yu, Y., Shen, M.,
Song, Q. & Xie, J. Biological activities and pharmaceutical applications of polysaccharide from natural resources: A review. _Carbohydr. Polym._ 183, 91–101 (2018). Article CAS PubMed
Google Scholar * Dashty, M. A quick look at biochemistry: carbohydrate metabolism. _Clin. Biochem._ 46, 1339–1352 (2013). Article CAS PubMed Google Scholar * Fadaka, A. et al. Biology
of glucose metabolization in cancer cells. _J. Oncol. Sci._ 3, 45–51 (2017). Article Google Scholar * Brown, G. D., Bauer, J., Osborn, H. M. & Kuemmerle, R. A solution NMR approach to
determine the chemical structures of carbohydrates using the hydroxyl groups as starting points. _ACS omega_ 3, 17957–17975 (2018). Article CAS PubMed PubMed Central Google Scholar *
Fels, L. & Bunzel, M. Application of accelerated heteronuclear single quantum coherence experiments to the rapid quantification of monosaccharides and disaccharides in dairy products.
_Magn. Reson. Chem._ 60, 692–701 (2022). Article CAS PubMed Google Scholar * Scarselli, F., Gori, M., Tsoi, A. C., Hagenbuchner, M. & Monfardini, G. The graph neural network model.
_IEEE Trans. Neural Netw._ 20, 61–80 (2008). Article PubMed Google Scholar * Micheli, A. Neural network for graphs: A contextual constructive approach. _IEEE Trans. Neural Netw._ 20,
498–511 (2009). Article PubMed Google Scholar * Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. In _International
Conference on Machine Learning_, 1263–1272 (PMLR, 2017). * Zhou, J. et al. Graph neural networks: A review of methods and applications. _AI Open_ 1, 57–81 (2020). Article Google Scholar *
Reiser, P. et al. Graph neural networks for materials science and chemistry. _Commun. Mater._ 3, 93 (2022). Article CAS PubMed PubMed Central Google Scholar * Edison, S. &
Schroeder, F. C. NMR – small molecules and analysis of complex mixtures. _Compr. Nat. Prod. II_ 9, 69–196 (2010). * Kuhn, H. W. The hungarian method for the assignment problem. _Nav. Res.
Logist. Q._ 2, 83–97 (1955). Article Google Scholar * Munkres, J. Algorithms for the assignment and transportation problems. _J. Soc. Ind. Appl. Math._ 5, 32–38 (1957). Article Google
Scholar * Gold, S. & Rangarajan, A. A graduated assignment algorithm for graph matching. _IEEE Trans. Pattern Anal. Mach. Intell._ 18, 377–388 (1996). Article Google Scholar Download
references ACKNOWLEDGEMENTS This work was supported by GlycoMIP, a National Science Foundation (NSF) Materials Innovation Platform funded through Cooperative Agreement DMR-1933525, NSF OAC
1920147, as well as NIH R24GM137782. AUTHOR INFORMATION Author notes * These authors contributed equally: Yunrui Li, Hao Xu. AUTHORS AND AFFILIATIONS * Department of Computer Science,
Brandeis University, Waltham, MA, USA Yunrui Li & Pengyu Hong * Department of Medicine, Harvard Medical School, Boston, MA, USA Hao Xu * Complex Carbohydrate Research Center, University
of Georgia, Athens, GA, USA Ambrish Kumar, Christian Heiss & Parastoo Azadi * Department of Chemistry, Boston College, Chestnut Hill, MA, USA Duo-Sheng Wang Authors * Yunrui Li View
author publications You can also search for this author inPubMed Google Scholar * Hao Xu View author publications You can also search for this author inPubMed Google Scholar * Ambrish Kumar
View author publications You can also search for this author inPubMed Google Scholar * Duo-Sheng Wang View author publications You can also search for this author inPubMed Google Scholar *
Christian Heiss View author publications You can also search for this author inPubMed Google Scholar * Parastoo Azadi View author publications You can also search for this author inPubMed
Google Scholar * Pengyu Hong View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Y. Li and H. Xu: idea generation, algorithm design, code
implementation, data analysis, manuscript writing, editing, and revision. A. Kumar and D. Wang: data annotation. C. Heiss and P. Azadi: supervision. P. Hong: supervision, research planning,
funding acquisition, manuscript review, editing and revision. All authors reviewed the manuscript. H. Xu’s work was done while he was at Brandeis University. CORRESPONDING AUTHOR
Correspondence to Pengyu Hong. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Communications Chemistry_ thanks the
anonymous reviewers for their contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 RIGHTS AND PERMISSIONS
OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution
and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you
modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in
this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Li, Y., Xu, H., Kumar, A. _et al._ TransPeakNet
for solvent-aware 2D NMR prediction via multi-task pre-training and unsupervised learning. _Commun Chem_ 8, 51 (2025). https://doi.org/10.1038/s42004-025-01455-9 Download citation *
Received: 30 October 2024 * Accepted: 11 February 2025 * Published: 20 February 2025 * DOI: https://doi.org/10.1038/s42004-025-01455-9 SHARE THIS ARTICLE Anyone you share the following link
with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative
Trending News
The bcl-2 pro-survival protein a1 is dispensable for t cell homeostasis on viral infectionABSTRACT The physiological role of the pro-survival BCL-2 family member A1 has been debated for a long time. Strong mRNA...
Small doses of alcohol affect driving skills | science newsScience News was founded in 1921 as an independent, nonprofit source of accurate information on the latest news of scien...
5 signs a house is a money pitThe spring home-buying season is in full swing, and if forecasts are correct it’s going to be a busy one. Despite mortga...
Drought dries up copper canyon waterfall although some blame miningA waterfall in the Copper Canyon in Ocampo, Chihuahua, has dried up due to the severe drought affecting the area. The 24...
Waste-to-biofuels technology ready for global rolloutBreakthrough technology developed by Enerkem converts a multitude of waste material into renewable fuel and chemicals, m...
Latests News
Transpeaknet for solvent-aware 2d nmr prediction via multi-task pre-training and unsupervised learningABSTRACT Nuclear Magnetic Resonance (NMR) spectroscopy is essential for revealing molecular structure, electronic enviro...
Bunge india set to revive palm oil brand lotusBunge India is resurrecting the Lotus brand of palm oil, 10 years after acquiring it along with the more popular Dalda, ...
Tracfone scores 215,000 customers during third quarter | rcr wireless newsMEXICO CITY-Prepaid wireless provider TracFone Wireless Inc. reported soaring third-quarter revenues of $330 million, a ...
New passover traditions emerge from pandemic yearAndrew Silow-Carroll, 60, editor of _The New York Jewish Week_, and his wife, Sharon, also had Zoom seders in 2020, with...
Prophylactic effects of amine oxides in radiation injury in miceABSTRACT IN 1957, Haley _et al._ 1 found that quinoxaline-1,4-di-N-oxide reduced X-radiation mortality in mice by 50 per...