Visualizing subcellular changes in the nad(h) pool size versus redox state using fluorescence lifetime imaging microscopy of nadh
Visualizing subcellular changes in the nad(h) pool size versus redox state using fluorescence lifetime imaging microscopy of nadh"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT NADH autofluorescence imaging is a promising approach for visualizing energy metabolism at the single-cell level. However, it is sensitive to the redox ratio and the total NAD(H)
amount, which can change independently from each other, for example with aging. Here, we evaluate the potential of fluorescence lifetime imaging microscopy (FLIM) of NADH to differentiate
between these modalities. We perform targeted modifications of the NAD(H) pool size and ratio in cells and mice and assess the impact on NADH FLIM. We show that NADH FLIM is sensitive to
NAD(H) pool size, mimicking the effect of redox alterations. However, individual components of the fluorescence lifetime are differently impacted by redox versus pool size changes, allowing
us to distinguish both modalities using only FLIM. Our results emphasize NADH FLIM’s potential for evaluating cellular metabolism and relative NAD(H) levels with high spatial resolution,
providing a crucial tool for our understanding of aging and metabolism. SIMILAR CONTENT BEING VIEWED BY OTHERS LABEL-FREE CHARACTERIZATION OF SINGLE EXTRACELLULAR VESICLES USING TWO-PHOTON
FLUORESCENCE LIFETIME IMAGING MICROSCOPY OF NAD(P)H Article Open access 08 February 2021 CHARACTERIZATION OF MITOCHONDRIAL DYSFUNCTION DUE TO LASER DAMAGE BY 2-PHOTON FLIM MICROSCOPY Article
Open access 13 July 2022 CONTRIBUTION OF AUTOFLUORESCENCE FROM INTRACELLULAR PROTEINS IN MULTIPHOTON FLUORESCENCE LIFETIME IMAGING Article Open access 05 October 2022 INTRODUCTION
Alterations in cellular energy metabolism and redox state are hallmarks of a variety of disorders such as cancer, diabetes, and neurodegenerative diseases1,2. In many such diseases,
metabolic alterations show tissue, cell, and even mitochondrial specificity3,4. Consequently, new approaches to evaluate metabolism with high spatial resolution are needed. One promising
method relies on the autofluorescence of the electron carrier nicotinamide adenine dinucleotide (NADH)5. Because glycolysis and the citric acid cycle reduce NAD+ to NADH, while the electron
transport system re-oxidizes NADH back to NAD+, the ratio of NAD+ to NADH can reveal shifts in cellular energy metabolism that favor mitochondrial respiration or glycolysis. Importantly,
NADH is autofluorescent while NAD+ is not, so higher NADH fluorescence intensity is associated with a more reduced redox state and vice versa6. Accordingly, it has been shown that blocking
mitochondrial respiration results in increased NADH intensity7 while blocking glycolysis results in lower NADH intensity8. As such, NADH imaging enables an estimation of cellular energy
metabolism and mitochondrial function microscopically, without external markers or reporters, which renders it an ideal candidate for in-vivo applications. To avoid drawbacks of intensity
imaging in tissues, such as differences in absorption or light scattering, NADH fluorescence lifetime imaging microscopy (NADH FLIM) can be used. NADH FLIM relies on the differences in the
fluorescence lifetimes between free NADH (around 400 ps) and protein-bound NADH (around 2500 ps)9. Since the protein-bound NADH pool remains relatively stable in comparison to the free NADH
pool, a larger protein-bound to free NADH ratio, or a longer mean NADH lifetime, indicates less NADH relative to NAD+ and a more oxidized redox state. Multiple studies have shown that
blocking mitochondrial respiration, which is expected to increase the free NADH pool, results in a shorter NADH lifetime10,11,12, establishing the use of NADH fluorescence lifetime as a
promising surrogate marker for cellular energy metabolism. However, NAD+ and NADH are involved in a multitude of redox reactions in the cell, meaning a variety of factors independent of
energy metabolism influence the detected NAD(H) redox state13. These include the pH, which is especially variable in the mitochondrial matrix10, and protein composition, which impacts the
lifetime of the protein-bound NADH14. Additionally, NADPH autofluorescence cannot be spectrally distinguished from NADH fluorescence15. As a result, these factors must be taken into account
when utilizing NADH imaging to evaluate cellular metabolism. NAD(H) pool size, defined as the total amount of NAD+ and NADH combined, is another variable that must be considered.
Importantly, NAD(H) pool size can be altered in many diseases, such as colon cancer16 and mitochondrial myopathy17. Likewise, aging is associated with decreased NAD+ levels and a decreased
NAD(H) pool due to dysfunction in NAD+ biosynthesis18,19. Accordingly, supplementation with NAD+ precursors, which not only affect NAD+ levels but to a lesser extend also NADH, has recently
gained much attention20. Thus, a non-invasive method to assess NAD(H) pool sizes in addition to redox state and metabolism would be extremely valuable. However, increases in NAD(H) pool size
cannot be distinguished from a more reduced redox state in NADH intensity imaging, since both cause an increase in fluorescence intensity. Thus far, though, the impact of changes in the
NAD(H) pool size on NADH lifetime from FLIM have not been investigated. Here, we hypothesize that combining NADH intensity and lifetime information using NADH FLIM can allow us to
simultaneously evaluate alterations in the NAD(H) pool and redox state, providing an extremely valuable tool, for example, to study aging. To test this, we perform targeted modifications of
NAD(H) pool size in cells and mice. We show that pool size is inversely correlated with the mean NADH lifetime, and subsequently establish different approaches to differentiate between pool
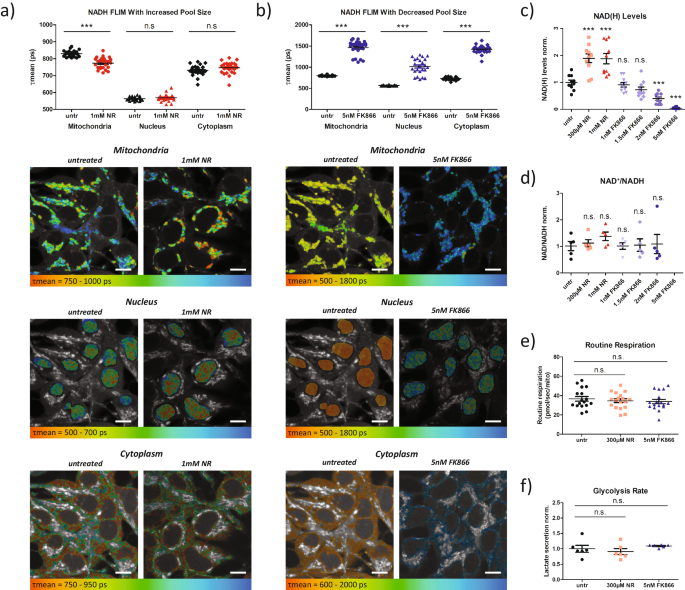
size-induced and respiration-induced changes in NADH autofluorescence. RESULTS CHANGES IN NAD(H) POOL SIZE IMPACT NADH FLIM To determine if NADH FLIM is sensitive to changes in NAD(H) pool
size, we first modified pool size by treating HEK293 cells with either nicotinamide riboside (NR), which feeds into the NAD+ salvage pathway, to increase pool size, or FK866, an NAD+ salvage
pathway inhibitor, to decrease pool size. Interestingly, treatment with NR caused a significant decrease in the mean NADH lifetime (τmean) in the mitochondria, but not the nucleus or
cytoplasm (Fig. 1a). Conversely, treatment with FK866 significantly increased τmean in the mitochondria, nucleus, and cytoplasm (Fig. 1b). These results suggest that the NADH lifetime is
sensitive to changes in the NAD(H) pool size. We next cross-validated these findings in 143B osteosarcoma cells to ensure that the effects of NAD(H) pool size on NADH FLIM are not cell type
specific. Similar to HEK293 cells, treatment with NR decreased mean NADH lifetime relative to control cells, and treatment with FK866 increased mean lifetime (Fig. S1a, b). Cytoplasmic NADH
was not analyzed separately since it equilibrates with the nucleus through the nuclear pores21. This was confirmed by correlating nuclear and cytoplasmic mean NADH lifetimes across our
treatment (Fig. S2). To validate that the observed changes in mitochondrial, nuclear, and cytoplasmic τmean upon NR and FK866 treatments were in fact due to alterations in NAD(H) pool size
and not due to changes in the redox ratio, we first assessed NAD+ and NADH levels biochemically. As expected, titrations of both NR and FK866 in HEK293 cells revealed dose dependent
alterations of NAD(H) pool size (Fig. 1c). Importantly, the biochemically quantified NAD+/NADH ratio was not significantly different for any condition (Fig. 1d), suggesting that neither
treatment strongly altered cellular redox state. Unfortunately, at 5 nM FK866, we could not assess the NAD+/NADH ratio reliably from the biochemical assays since NAD(H) levels were too low.
Thus, to avoid bias, we performed NADH FLIM of HEK293 cells treated with varying concentrations of FK866 and NR, which showed that FK866 increased τmean and NR decreased τmean in a
concentration dependent manner (Fig. S3a, b). To further verify that neither treatment significantly impacted NADH lifetimes by modifying energy metabolism, we performed Oroboros high
resolution respirometry on cells treated with 300 µM NR and 5 nM FK866. We revealed that routine respiration, which mirrors the respiratory state during FLIM measurements, was not altered
(Fig. 1e). Likewise, electron transport system capacity (ETS capacity), leak respiration, and the respiratory control ratios were not significantly altered, and neither was doubling time
(Fig. S3c–h). Additionally, we measured lactate secretion rate. Similar to respiration, treated cells showed no significant difference in lactate secretion compared to controls (Fig. 1f),
suggesting no change in glycolysis. Finally, to additionally confirm neither NR nor FK866 impact metabolism, we performed Seahorse metabolic flux assays on 143B and HEK293 cells treated with
NR and FK866 to determine basal respiration. Oxygen consumption rate (OCR) was not effected by either treatment in 143B cells (Fig. S4a), but 5 nM FK866 treatment significantly lowered OCR
in HEK293 cells (Fig. S4b, c). This was only observed at the highest FK866 concentration, and is not totally surprising given that that 5 nM FK866, NAD(H) levels are nearly undetectable
(Fig. 1c). The OCR/ECAR ratio was not impacted by either treatment in 143B cells (Fig. S4d), but was decreased in HEK293 cells treated with FK866 (Fig. S4e, f). Thus, FK866 treatment shifts
metabolism in HEK293 cells towards glycolysis. However, this cannot explain the lifetime changes we observe, because a decreased OCR/ECAR ratio suggests a more reduced redox ratio, which
would shorten the NADH lifetime. Instead, we observe significantly increased lifetime upon FK866 treatments, recapitulating that the altered lifetimes upon NR and FK866 treatment are not
caused by differences in cellular energy metabolism. Since NADPH autofluorescence cannot be distinguished from NADH fluorescence spectrally, we also quantified NADP(H) levels biochemically.
As expected, NADP(H) levels follow similar trends as NAD(H) levels upon NR and FK866 treatments (Fig. S5a,b). Comparing the relative amounts of NADPH versus NADH revealed that NR treatment
did not change the ratio of NADPH to NADH, but FK866 treatment resulted in higher NADPH/NADH ratio (Fig. S5c). To verify our biochemical quantifications, we also performed analysis developed
by Blacker et al. to determine the NADPH/NADH ratio in treated and untreated cells using lifetime components from FLIM15 (Fig. S5d, e). This analysis showed close correlations between the
optically quantified NADPH/NADH ratio in both mitochondria and the nucleus and whole cell biochemical quantifications of NADPH/NADH, suggesting no significant differential effects on the
subcellular level (Fig. S5f, g). To confirm this, we isolated cytosolic and mitochondrial fractions from NR and FK866 treated cells, and biochemically quantified NADPH/NADH in each
subcellular fraction and in whole cell samples collected prior to fractionation (Fig. S5h). In accordance with prior studies, we found that NADPH/NADH is higher in cytosol than in
mitochondria22. Crucially though, treatment with both FK866 and NR has a similar effect on NADPH/NADH in mitochondria and in cytosol compared to whole cells, reiterating that neither NR or
FK866 causes differential changes to the NADPH/NADH ratio across cellular compartments. Thus, their impact on subcellular NADH FLIM results is also minimal. Given that NADPH has been
reported to have a longer protein-bound lifetime compared to NADH15, the increased fraction of NADPH might, in part, explain the longer mean NAD(P)H lifetime we observed upon FK866
treatment. However, given that FK866 affected nuclear and mitochondrial τmean similarly, although the NADPH to NADH ratio is known to typically be higher in nuclei compared to mitochondria,
it is highly unlikely that the entire FK866 effect is mediated by higher NADPH/NADH. Similarly, the NAD(P)H lifetime alterations upon NR treatment cannot be explained by changes in NADPH.
Taken together, our results demonstrate, that in contrast to expectations, changes in the NAD(H) pool size do not only affect the NADH autofluorescence intensity but also the mean NADH
lifetime, resulting in an inverse relationship between pool size and mean NADH lifetime. NADH FLIM ALLOWS VISUALIZATION OF NADH POOL SIZE DIFFERENCES ON A SINGLE CELL LEVEL To demonstrate
that NADH FLIM can be used to evaluate NAD(H) pool size or redox changes even on a single cell level, we expressed mitochondrially targeted _Lactobacillus brevis_ H2O-forming NADH oxidase
(mt_Lb_NOX)23 in cells. We created a bicistronic plasmid which expressed the fluorescent protein CayenneRFP and mt_Lb_NOX to similar levels. This allowed us to estimate mt_Lb_NOX expression
by measuring fluorescence intensity of CayenneRFP in each cell. Stable cell lines were established without clonal selection, resulting in heterogeneous expression of mt_Lb_NOX across
individual cells. First, we biochemically assessed the effect of mt_Lb_NOX expression on both NAD+ and NADH levels in 143B osteosarcoma cells (Fig. 2a–c). Contrary to Titov et al.’s findings
in their original paper characterizing mt_Lb_NOX expressing cells23, we found a significant decrease in both NAD+ and NADH levels (Fig. 2d, e). Subsequent testing with an alternate
biochemical quantification kit confirmed these findings and demonstrated no significant difference in NAD+/NADH ratio between control and mt_Lb_NOX expressing cells (Fig. S6a–c_)_, possibly
because the cell naturally equilibrates the redox state even when only NADH is scavenged by mt_Lb_NOX. To test this hypothesis, we quantified NAD+ and NADH one week and two weeks (data in
Fig. S6a–c) after starting selection pressure with G418 in our mt_Lb_NOX cells (Fig. S6d–f). At the earlier 1-week time point, the redox ratio is more reduced in mt_Lb_NOX expressing cells,
which more closely matches Titov et al.’s results23. However, NAD+ levels were already significantly decreased, suggesting that the cells had begun to equilibrate their redox ratio. These
results demonstrate that the time after transfection and the respective cellular response to _Lb_NOX expression play a significant role in the effect of _Lb_NOX on NAD(H) pool size and redox
ratio, and suggest that our mt_Lb_NOX overexpressing cells can be used as an appropriate tool to study the effect of a reduced NAD(H) pool size but stable redox ratio on NADH FLIM. Because
mt_Lb_NOX expression varied between cells, the mean NADH lifetime in the nucleus and mitochondria was quantified for each individual cell. Simultaneously, CayenneRFP fluorescence intensity
was measured as a surrogate marker of mt_Lb_NOX expression of each cell. Next, we correlated the mean NADH lifetime to mt_Lb_NOX expression, revealing a positive correlation for
mitochondrial τmean (Fig. 2a). No correlation was seen for nuclear or cytoplasmic τmean (Fig. 2b, c), consistent with the mitochondrial localization of mt_Lb_NOX. This can be seen in
side-by-side comparisons of CayenneRFP fluorescence intensity images (Fig. 2a–c, right images) and false-color coded images of the mean NADH lifetime (Fig. 2a–c, left images). Cells with
brighter CayenneRFP fluorescence clearly showed longer mitochondrial NADH lifetimes (top left image, more blue) but no alteration in the nuclear or cytoplasmic NADH lifetime (middle and
bottom left images). The lack of alterations in nuclear and cytoplasmic NADH lifetime additionally confirmed that our expression marker, Cayenne RFP, was not impacting NADH lifetime
measurements. Since mt_Lb_NOX itself consumes oxygen23, standard respirometry could not be performed to rule out changes in cellular energy metabolism. To validate that the observed
variation in NADH lifetimes between high and low mt_Lb_NOX expressing cells was not due to differences in metabolism, cells were treated with the respiratory chain inhibitor antimycin A
(AA)10. By eliminating respiration in all cells independent of their mt_Lb_NOX expression levels, any remaining correlation between mt_Lb_NOX and NADH lifetime could not be due to
differences in mitochondrial respiration. Indeed, we still observed a significant correlation between mt_Lb_NOX expression and mitochondrial NADH lifetime (Fig. 2a, red line) but not in the
nucleus or cytoplasm (Fig. 2b, c, red line). Furthermore, we detected no differences in the slopes of the correlations before and after antimycin A treatment (Fig. 2a–c, red and black
lines), indicating that the effect of mt_Lb_NOX on the mitochondrial lifetime is not mediated by mt_Lb_NOX altering mitochondrial respiration and redox state. Next, to rule out the
possibility that binding of NADH to mt_Lb_NOX was impacting the protein-bound NADH fluorescence lifetime, we plotted τ2, representative of the protein bound NADH lifetime, against the
mt_Lb_NOX expression (Fig. 2f). Unlike τmean, τ2 showed no significant correlation with mt_Lb_NOX expression, suggesting that mt_Lb_NOX expression does not significantly impact the
protein-bound NADH lifetime. To sum up, we demonstrated that NADH FLIM can visualize pool size differences on a single cell level between adjacent cells in a heterogeneous population, and
further validated that pool size and mean NADH lifetime are inversely correlated. NADH FLIM CAN DETECT CHANGES IN NADH POOL SIZE IN TISSUES NR has been proposed as treatment for a large
number of pathological conditions and age related diseases by increasing NAD+ levels or NAD(H) pool size20. To assess whether pharmacological modifications to the NAD(H) pool size in-vivo
can be visualized within tissues, mice were injected with 500 mg/kg NR or 0.9% NaCl. As the NAD+ salvage pathway is most prominent in the liver, liver tissue was dissected and imaged three
hours after mice were injected with treatments. When performing NADH FLIM of liver tissue, we observed bright spots of autofluorescence in our NADH channel (emission 430–490 nm) that
demonstrated a short lifetime component of around 1000 ps (Fig. S7a, right images). This signal was present in all mice, regardless of genotype or treatment group, and interestingly, seemed
to increase in frequency but not intensity with age (Fig. S7a, lower vs. upper images). As 1000 ps is not a typical NADH fluorescence lifetime component, we hypothesized that these bright
spots were due to non-NADH autofluorescent molecules. This was confirmed by the absence of a response of this signal to antimycin A, which blocks respiration and thereby reduces the NADH
lifetime10. In the phasor plot, a graphical representation of fluorescence lifetime characteristics, antimycin A treatment caused a shift in the population of pixels that represented true
NADH signal in the phasor plot, but no shift in the unknown population, confirming that the bright spots did not represent NADH (Fig. S7b). Thus, in subsequent analyses, the region on the
phasor plot which corresponded to NADH fluorescence was selected to exclude non-NADH fluorescence (Fig. S7c). Given the fluorescence characteristics of this unknown signal and its increase
with age, we hypothesize that it might represent lipofuscin. As expected, liver samples from mice treated with NR showed a shorter mean NADH lifetime (Fig. 3a). Interestingly, we observed
strong regional differences in the mean NADH lifetime within individual liver pieces. This might be due to metabolic differences within liver lobules, suggesting NADH FLIM can be a promising
technique to reveal these metabolic differences in future studies. To verify the effect of NR on liver nucleotide levels, we quantified NAD+ and NADH. Indeed, NR increased liver NAD+ and
NADH levels to a similar extent (Fig. 3b), demonstrating an increased NAD(H) pool size but no redox changes. Overall, these data demonstrate that changes in NAD(H) pool size directly result
in inverse changes in the mean NADH lifetime across different cell lines as well as in tissues. On a positive note, this allows assessment of the NAD(H) pool size optically with high spatial
resolution using NADH FLIM. On the other hand, it complicates the interpretation of the mean NADH lifetime as a marker for either energy metabolism, NAD(H) redox state, or NAD(H) pool size
alone. Thus, approaches must be established to distinguish the underlying biological modalities that can affect the mean NADH lifetime. INHIBITION OF RESPIRATION ENABLES SEPARATION OF
RESPIRATION INDUCED AND NON-RESPIRATION INDUCED NADH FLIM CHANGES Thus far, we have shown that NAD(H) pool size impacts NADH FLIM independent of respiration. However, when NADH FLIM is being
used as an indirect marker of cellular energy metabolism, simultaneous alterations in the NAD(H) pool size can confound those measurements. Thus, in order to evaluate metabolic changes, it
is essential to isolate respiration-induced changes in NADH FLIM from changes induced by other factors. To accomplish this, cells were treated with antimycin A, a complex 3 inhibitor,
immediately before the FLIM measurement. This eliminates any changes in the mitochondrial redox ratio due to respiration but does not impact many other factors, like total cellular NAD(H)
pool size, which may confound the FLIM measurements when assessing cellular metabolism. Thus, if a difference between treatment and control persists after AA treatment, it is not due to
respiration. We applied this concept to NR and FK866 treated 143B and HEK293 cells, of which we know NAD(H) pool size is altered (Fig. 1c), while respiration remains largely unchanged (Fig.
1e). As demonstrated previously (Fig. 1, Fig. S1, Fig. S3), NR resulted in a shorter and FK866 in a longer mitochondrial mean NADH lifetime. These differences persisted upon treatment with
AA (Fig. 4a, c), demonstrating again that the differences in NADH lifetime between NR, FK866, and untreated cells cannot be due to differences in respiration, since mitochondrial respiration
was zero across all conditions following AA. By calculating the difference between the mean NADH lifetime with and without respiration-inhibition, which we term metabolic delta, the impact
of respiration on NADH FLIM can be isolated in a single parameter. Similar metabolic delta values between populations suggest similar levels of respiration, regardless of the absolute mean
lifetime values. Performing metabolic delta calculations on the NR and FK866 treated 143B cells revealed similar metabolic delta values for all 3 treatments (Fig. 4b), reiterating that the
changes we see in NADH FLIM after NR and FK866 treatment are not due to metabolic changes. However, metabolic delta of NR treated HEK293 cells was slightly lower than untreated controls
(Fig. 4d). This difference was significant, despite no change in mitochondrial respiration (Fig. 1e), and the same trend was observed in 143B cells, although it did not reach significance.
To further understand this limitation, we plotted the total cellular NAD(H) levels after NR and FK866 treatment against the respective average τmeans (Fig. S8a). This correlation
demonstrates that the relationship between NAD(H) pool size and mean NADH lifetime is not completely linear. In fact, a 50% increase in NAD(H) pool size compared to untreated controls
resulted in a 50 ps decrease in mean NADH lifetime. In comparison, a 50% decrease in NAD(H) pool size resulted in a nearly 150 ps increase in mean NADH lifetime. This is likely because the
mean NADH lifetime is limited by the protein bound NADH lifetime of 2500 ps and the free NADH lifetime of 400 ps. Since 70–80% of the NADH is free in the untreated state, further increases
in free NADH due to the reduced redox change induced by AA causes the mean lifetime to change less the closer it is to its lower limit of 400 ps. Since NR increases NAD(H) levels, causing
the mean NADH lifetime to decrease towards the lower limit, metabolic delta for NR treated cells is consequently lower than that of untreated and FK866 treated cells. These results highlight
one limitation of the metabolic delta as an indicator of respiratory differences upon drastic NAD(H) pool size changes. A further limitation is that the metabolic delta can only eliminate
the impact of factors which are stable upon acute AA treatment. Interestingly, we revealed that the nuclear mean NADH lifetime is significantly increased upon acute AA treatment (Fig. S8b,
c), suggesting a more oxidized NAD+/NADH ratio. However, this is opposite to what would be expected biologically, since AA should result in an increase in glycolysis and reduces the pyruvate
to lactate ratio (Fig. S8d), a known marker for the cytosolic NAD+/NADH ratio24. A possible explanation is that because antimycin A inhibits respiration and complex I is unable to
re-oxidize NADH back to NAD+, NAD+ is transported into the mitochondria through the NAD+ transporter SLC25A5125. This would cause a local decrease in NAD(H) pool size in the nucleus,
resulting in the longer NADH lifetime upon AA treatment. This emphasizes the critical role of changes in the NAD(H) pool size not only on the cellular level, but also between subcellular
compartments. It further demonstrates that subcellular pools of NAD(H) are NOT stable upon acute AA treatment, rendering metabolic delta an imperfect surrogate marker for mitochondrial
respiration, although its performance is still superior to using the mean NADH lifetime alone (compare Fig. 4a, c versus Fig. 4b, d). ANALYSIS OF FLIM COMPONENTS ALLOWS DIFFERENTIATION OF
POOL SIZE AND RESPIRATION INDUCED LIFETIME CHANGES To overcome the limitations of the metabolic delta mentioned above, as well as its limited applicability in tissues and in-vivo, we asked
if specific changes in individual components of the mean NADH lifetime are associated with pool-size induced or respiration-induced alterations in τmean. Thus, we re-examined NADH FLIM data
from mt_Lb_NOX treated cells and untreated/antimycin A treated cells because the variance in τmean between mt_Lb_NOX treated cells is solely due to differences in the NAD(H) pool size, while
the variance in τmean in untreated vs antimycin A treated cells is due to differences in respiration. To assess how the individual lifetime components contribute to changes in τmean, we
then plotted them against τmean. We showed that respiration-induced changes in τmean are not associated with changes in the τ1 component (free NADH lifetime) while pool size-induced
increases in τmean are associated with a parallel increase in the τ1 component (Fig. 5a). In contrast, respiration-induced changes in τmean are mainly driven by an altered ratio of free (a1)
to protein-bound (a2) NADH (a1/a2), while this change in a1/a2 is less prominent for pool size-induced alterations of τmean (Fig. 5b). To verify these observations upon different treatment
conditions, we performed similar analyses in 143B cells between FK866 and NR treatments (pool-size induced) and between untreated and antimycin A treatments (respiration-induced). We
calculated the fold change of each NADH FLIM component normalized to the change in τmean. Similar to our previous results, pool size-induced changes in τmean were associated with a larger
fold change in τ1 and τ2 but smaller changes in free (a1abs), protein bound (a2abs) and total (aabs) NADH autofluorescence intensities and the ratio of free to protein bound NADH (a1/a2)
compared to respiration-induced changes in τmean (Fig. 5c). This was further validated in HEK293 cells, which also demonstrate that pool-size induced changes in τmean are driven by changes
in τ1, while respiration-induced changes in τmean are driven by changes in a1/a2 (Fig. S9). Although the exact mechanism behind these differential changes are not fully elucidated, the
consistency of our observations across 2 cell lines and with both chemical (NR/FK866) and enzymatic (mt_Lb_NOX) modifications of NAD(H) pool size, provides strong descriptive evidence for
the validity of these observations. Consequently, individual lifetime components, in particular τ1 and a1/a2, can be utilized to predict whether an observed difference in the mean NADH
lifetime between two conditions is due to NAD(H) pool size differences (strong difference in τ1) or due to differences in mitochondrial respiration/redox ratio (strong difference in a1/a2)
(Fig. 6). This highlights the potential of NADH FLIM as a tool to evaluate both NAD(H) redox state and pool size changes with subcellular resolution, providing unique insights into redox
metabolism in health and disease. DISCUSSION Here, we performed targeted modifications of NAD(H) pool size and redox state and assessed the extent to which fluorescence lifetime imaging
microscopy of NADH autofluorescence can detect and distinguish these modifications in cells and tissues. This provided three crucial insights in NADH autofluorescence imaging and its
potential in bioenergetic research. First, we demonstrated that NAD(H) pool size is inversely related to the mean NADH lifetime, so a smaller NAD(H) pool size results in a longer mean NADH
lifetime and vice versa. Thus, NADH FLIM can be used to evaluate NAD(H) pool size in cells and tissues when redox state is stable. While NADH autofluorescence intensity is known to be
sensitive to NAD(H) pool size and has previously been used to estimate NAD(H) pool size16, NADH FLIM relies on the fraction of free to protein-bound NADH and their individual lifetimes τ1
and τ2, which are dependent on the respective microenvironment9. Thus, as a ratio-metric readout, one might have hypothesized that NADH FLIM is insensitive to alterations in NAD(H) pool
size. However, we demonstrated a causal influence of NAD(H) pool size on the mean NADH lifetime. This can be explained by earlier observations that the protein-bound fraction of NADH is more
stable than the free NADH fraction14. Thus, an overall decrease in NAD(H) pool size does not cause an equivalent decrease in the free NAD(H) pool compared to the protein-bound NAD(H) pool.
Instead, the free NAD(H) pool will show a much larger decrease, thus also affecting the mean NADH lifetime. Consequently, the mean NADH lifetime from NADH FLIM appears to reflect NADH
concentration similar to intensity imaging, whether it is affected by changes in redox ratio or NAD(H) pool size. This raises the question of the additional value of the more technically
complex NADH FLIM as opposed to NADH intensity imaging. Most importantly, FLIM imaging is independent of the recorded fluorescence intensity26, so the results are not affected by differences
in absorption or light scattering, which is particularly crucial when imaging deep in tissues or in vivo. Furthermore, by providing the fluorescence intensities of free and protein-bound
NADH and their respective lifetimes, FLIM reveals additional information that can be utilized in the biological interpretation of the data, for example for revealing the fraction of NADPH27
or changes in the proteins which NADH binds to14. For evaluation of pool size, compared to the current standard of biochemical assays detecting NAD+ and NADH28, NADH FLIM is non-destructive,
and more importantly, allows visualization of pool size differences on a single cell or even subcellular level, as demonstrated by our imaging of mt_Lb_NOX expressing cells (Fig. 2a–c).
Similarly, genetically-encoded biosensors, such as SoNar and iNap, also allow subcellular visualization of NAD(P)(H)29,30. The advantage of the biosensors is a high specificity for NADH over
NADPH (or vice versa) as well as an easier calibration for comparison of absolute redox ratios across cell compartments/cell types. However, NADH FLIM does not require any additional
cellular manipulations to express NAD(H) biomarkers, which may be challenging in sensitive or difficult to transfect cell lines. This also means that, unlike genetically encoded biosensors,
FLIM can be used directly to quantify NAD(H) pool in tissues, as we have demonstrated in Fig. 3, or even humans31. Given the prominent role of alterations in nucleotide levels with aging and
disease32, NADH FLIM can be an invaluable tool in the study of a multitude of disorders. For example, increases in pool size have been associated with colorectal cancer progression16. Thus,
NADH FLIM can provide a non-invasive method to assess cancer progression33. Likewise, NADH FLIM has been used to non-invasively distinguish alterations in cerebral metabolism in live
rats34. A similar approach can be applied to assess changes in the NAD(H) pool size, for example in neurodegenerative diseases like Alzheimer’s disease35. This emphasizes the potential of
NADH FLIM in estimating alterations in NAD(H) nucleotide levels with subcellular resolution and in vivo. Second, our finding that NAD(H) pool size impacts the mean NADH lifetime highlights
the importance of considering NAD(H) pool size when estimating energy metabolism using NADH FLIM. The most common application for NADH FLIM is assessing energy metabolism and redox state, in
particular, the balance of mitochondrial respiration to glycolysis36. Since aging and age-associated disorders are associated with metabolic alterations37, multiple studies have utilized
NADH FLIM to assess metabolism upon aging38,39. Although mitochondrial function is known to decline with age40, some studies report a lower free to protein-bound NADH ratio (analogous to a
longer τmean) in aged vs. young mice38. Traditionally, a longer mean NADH lifetime is interpreted as more oxidative metabolism since it suggests more NAD+ relative to NADH11,41. However, it
is unlikely that aged mice are in fact performing more OXPHOS than young mice. Instead, the longer mean NADH lifetime may be due to a decrease in NAD(H) pool size in tissues of the aged
mice, consistent with the reported decline of NAD+ biosynthesis with age42. This emphasizes that potential changes in the cellular NAD(H) pool size need to be considered to avoid
misinterpretations of NADH FLIM results with respect to energy metabolism. Another scenario when pool size changes might have masked changes in redox state is upon NR treatment. Hu et al.
reported that NR causes an increase in cytosolic, but not mitochondrial NAD+/NADH ratio43. We have observed a significant decrease in NADH lifetime in the mitochondria but not the
nucleus/cytosol (Fig. 1a). The more oxidized redox state in the cytosol would increase the NADH lifetime, while the increased NAD(H) pool would decrease the NADH lifetime, resulting in no
effect of NR on the cytosolic τmean. In contrast, in the mitochondria the NAD+/NADH ratio does not change but the NAD(H) pool is increased, causing a shorter NADH lifetime. This highlights
the strong interconnection of both redox state and NAD(H) pool size in altering the NADH fluorescence signal. Interestingly, the effect of NAD(H) pool size on NADH FLIM does not only come
into play in case of systemic alterations in nucleotide levels. Recently, a mammalian mitochondrial NAD+ carrier was identified suggesting active redistribution of nucleotides pools between
mitochondria and cytosol25. Accordingly, it has been shown that the subcellular distribution of NAD(H) changes with cellular differentiation44 and metabolic state45. Thus, the local NAD(H)
pool size within cellular compartments can change. Here we have demonstrated how this can result in a misleading interpretation of NADH FLIM. We observed a longer nuclear NADH lifetime
following acute antimycin A treatment in both HEK293 and 143B cells (Fig. S8b, c), which normally would be interpreted as a more oxidized nuclear and cytosolic redox state. However,
antimycin A blocks respiration thereby increasing glycolysis and shifting the cytosolic NAD(H) redox state towards a reduced state, verified by a reduced pyruvate to lactate ratio. The most
likely explanation for a longer nuclear NADH lifetime despite a more reduced redox state is that blockage of the ETC causes nuclear NADH to be shuttled to and trapped in the mitochondria,
where it is unable to be re-oxidized. This would cause a local decrease in NAD(H) pool size in the nuclei, accounting for the increased mean NADH lifetime upon AA treatment. Overall, this
highlights that the effect of NAD(H) pool size on NADH FLIM always has to be considered when interpreting NADH FLIM data with respect to cellular metabolism or redox state. Finally, we
developed two approaches to distinguish pool size-induced and respiration-induced changes in NADH FLIM. Such separation is important for interpretation of NADH FLIM data because the two
scenarios have differing cellular implications. A longer mean lifetime due to changes in respiration suggests increased oxidative phosphorylation and a more oxidized NAD+/NADH ratio. This
impacts the balance of all cellular reactions that are dependent on the redox ratio, such as the TCA cycle dehydrogenases. On the other hand, a longer NADH lifetime due to pool size-induced
changes suggests an overall decrease in NAD(H) levels, which mostly affects reactions that require NAD+ as a cofactor, such as sirtuins. For example, a longer lifetime due to changes in
respiration would indicate more NAD+ relative to NADH and increased (or unaltered) sirtuin activity46. On the other hand, a pool size-induced increase in NADH lifetime would suggest
decreased NAD+ (and NADH) levels, resulting in lower sirtuin activity. This highlights how a similar effect on the mean NADH lifetime can encode opposing biological underpinnings,
emphasizing the need for further interpretation of the mean NADH lifetime. To isolate respiration-induced changes, we blocked mitochondrial respiration with antimycin A and calculated the
difference in NADH lifetime between untreated and respirationally-blocked cells, termed metabolic delta4. This allowed correction for all factors which remain stable upon inhibition of
respiration. For example, the total cellular NAD(H) pool size does not change with an acute treatment of antimycin A. Thus, a larger cellular NAD(H) pool size will affect the mean NADH
lifetimes of untreated and antimycin A -treated cells similarly, rendering the metabolic delta largely insensitive to alterations in the NAD(H) pool size. However, this approach has two
limitations. First, we demonstrated that τmean is not linearly correlated with NAD(H) pool size when τmean approached the free and protein bound lifetime limits of 400 ps and 2500 ps. Since
the fractions are usually shifted towards free NADH41, this results in slightly lower metabolic deltas when the overall NADH lifetimes are shorter. Accordingly, we observed a slightly lower
metabolic delta in NR treated cells compared to FK866-treated cells, although mitochondrial respiration was largely comparable. The same applies for the correlation of τmean with
respiration. However, metabolic changes only induce 100-200 ps differences in τmean when factors such as pool size and pH are controlled for. Thus, the relationship between τmean and
respiration is more likely to be monotonic in the relevant ranges10. Second, we demonstrated that, while acute antimycin A treatment does not alter total cellular NAD(H) pools, it shifts the
intracellular NAD(H) localization away from the nucleus and towards the mitochondria. Thus, when performing subcellular analysis, intracellular NAD(H) pool size may not be stable and can
impact FLIM results. Therefore, the metabolic delta provides a decent but still imperfect estimate of the contribution of respiration-induced NADH lifetime changes. Our second approach was
based on the hypothesis that individual parameters of NADH FLIM are differentially sensitive to respiration-induced versus pool size-induced alterations in the mean NADH lifetime. Indeed, we
could demonstrate that respiration-induced lifetime alterations are mostly due to changes in the ratio of free to protein-bound NADH (a1/a2), whereas pool-sized induced alterations show
significantly higher changes in τ1 and partially in τ2. One underlying reason for these different characteristics could be that changes in the NAD(H) pool size affect NADP(H) pool size to a
lesser extent, altering the ratio of NADPH to NADH. It has been reported that protein-bound NADPH has a longer lifetime compared to NADH15. Thus, the change in the NADPH to NADH ratio could
explain the influence of τ2 on pool-size induced lifetime alterations. However, the free NADPH lifetime is identical to free NADH lifetime15, rendering it unlikely that alterations in
NADPH/NADH are the main contributing factor for the overproportional changes in τ1 with alterations in NAD(H) pool size. Since the lifetime components τ1 and τ2 are sensitive to the
microenvironment, such as viscosity and pH14,47, we hypothesize that changes in pool size result in a different subcellular distribution of NAD(H) compared to changes in respiration, causing
differential changes in the cellular microenvironment. This results in specific changes in FLIM parameters, allowing differentiation of respiration-induced NADH lifetime alterations from
pool size-induced alterations. Although the exact underlying molecular mechanisms behind these observations are not fully elucidated, they are consistent across 143B cells and HEK293 cells,
as well as with 2 different methods of altering NAD(H) pool (chemically via NR/FK866 and enzymatically via mt_Lb_NOX), providing strong descriptive evidence that the differential changes are
genuine. However, it is possible that our findings may not be corroborated in different cell types or in tissues, thus, more work in the future is needed to determine whether FLIM
parameters behave similarly across cell types and tissues upon targeted modifications to respiration or NAD(H) pool size. Despite this, our work depicts an important stepping stone in first
identifying the NAD(H) pool size as a factor that should be considered in NADH FLIM interpretations and providing a working hypothesis to differentiate the influencing factors. The
differential changes in FLIM parameters with pool size vs respiration changes also inform additional methods for analyzing NADH FLIM data depending on the selected application. For example,
when detection of respiration changes is desired, metabolic delta can be calculated using the ratio of free to protein-bound NADH (a1/a2) rather than τmean, as we show that a1/a2 is less
sensitive to pool size alterations than τmean48. Thus, the resulting values are less likely to be confounded by any pool size changes. Overall, given that NADH autofluorescence is only a
surrogate marker, additional measures may be needed to ensure its accurate interpretations with respect to energy metabolism, redox state and NADH pool size. Firstly, NADH FLIM of antimycin
A treated samples enables correction for a range of different factors, including NAD(H) pool size to some extent, and can isolate changes in respiration between samples. Next, when using
FLIM as a direct measure of mitochondrial respiration, a matrix pH sensor may need to be simultaneously imaged to correct for pH mediated redox changes, as matrix pH tends to fluctuate
greatly10. Finally, to ensure that NAD(H) pool size is not a confounding factor, separate samples can be saved for biochemical quantification of total NAD(H). However, because this does not
address cases of re-distribution of NAD(H) within the cell, fold change analysis of the FLIM parameters can optionally be performed to further distinguish between respiration-induced and
NAD(H) pool size-induced lifetime changes. In summary, we reveal that NADH FLIM is sensitive to changes in the levels of NAD+ and NADH, promoting NADH FLIM as a valuable tool to estimate
NAD(H) pool size optically. At the same time, our findings reveal NAD(H) pool size as a crucial parameter which needs to be considered when performing NADH FLIM to evaluate cellular energy
metabolism and mitochondrial function. Despite this, we are able to use additional analytical approaches to estimate mitochondrial respiration, redox state, and NAD(H) pool size with
subcellular resolution, highlighting the potential of NADH FLIM as a crucial tool in aging and bioenergetics research. METHODS MOUSE STRAINS AND NR TREATMENT All mice used in this study are
male on the C57BL/6Eij (Nnt + /+) background (“B6” controls). In addition, mice deficient for the adenine nucleotide translocator 1 (ANT1, Slc25a4-/-)49 were used that further harbor the
complex I variant ND5 m.12352 C > T (ND5S204F). All mice were housed in a barrier facility, fed a low-fat 5L0D diet and kept on a 12 h light/dark cycle. Only male mice between 6 and 24
months of age were used in this study. Since the goal of the study is the methodological establishing of NADH FLIM in liver tissue and not the underlying biology, we did not assess both
genders. For NR treatment, NR (Nicotinamide Riboside chloride) powder (Chromadex) was dissolved in in water (15 mg/ml) and sterile filtered on the day of the experiment. The NR solution or
0.9% saline solution (sham) were IP-injected into the mice (100 μl/30 g of mouse weight) to achieve a final NR concentration of 500 mg/kg body weight. Animals were sacrificed by cervical
dislocation 3 h post injection, the right liver lobe was dissected, parts of it flash frozen and stored at –80 °C for biochemical analysis and one part imaged using NADH FLIM. The
Institutional Animal Care and Use Committee from the Children’s Hospital of Philadelphia approved all animal protocols of this research. We have complied with all relevant ethical
regulations for animal use. CELL CULTURE 143B(TK-) osteosarcoma cells and HEK293 cells were maintained in T75 cell culture flasks in 10 ml of Dulbecco’s modified medium (DMEM) from Gibco
(10569-010) supplemented with 10% fetal bovine serum and 2 mM uridine at standard cell culture conditions (37 °C, 5% CO2). 143B(TK-) cells were obtained from the Wallace laboratory
collection of cell lines, and HEK293 cells obtained from the American Type Culture Collection. Cell identities were confirmed by karyotype analysis and cell lines routinely screened for
mycoplasma using commercial PCR kits. Cells were split every 2-3 days. For the experiments, cells were seeded 48 h prior and the medium was changed 24 h prior to the experiment. NR and FK866
treatments were administered with the fresh media. NR-containing media was prepared fresh every week to exclude a change in concentration due to possible degradation. OROBOROS
High-resolution respirometry was performed using the Oroboros Oxygraph-2k in intact cells to mirror the energy metabolic state (endogenous substrates) during NADH FLIM10. Oroboros chambers
were first calibrated to room air using 2 mL of cell culture media (as described above). Conditioned media from the cells was collected and spun to remove cells and debris (1000 g, 3 min).
Cells were washed with PBS, trypsinized and resuspended in their conditioned media at a concentration of 1 million cells per mL. 2 million cells were loaded into each chamber, and the
oxygraph signal was allowed to plateau. This plateau represented routine respiration. Subsequently, 0.5 μL of 10 µM oligomycin was added to block Complex 5 activity and determine leak
respiration. Next, 1 μL of 1 mM FCCP was added to uncouple the inner mitochondrial membrane and simulate maximal ETC activity. After the signal plateaued, additional FCCP was added in 0.5 μL
increments until no further increase in respiration was observed. This final plateau corresponded to ETS capacity. Finally, 2 μL of 5 mM antimycin A was added to inhibit complex 3 and
measure background or non-mitochondrial oxygen consumption. GLYCOLYSIS AND DOUBLING TIME Conditioned media was collected in the context of the Oroboros measurements and stored at –80 °C
until lactate levels were measured using the Glycolyis Cell-Based Assay Kit (Cayman Chemicals). Lactate levels were normalized to the collection time and average cell number during the
collection time to reveal the lactate secretion rate50. Doubling time was calculated by counting cells prior to plating and again when harvesting the cells for the Oroboros experiments. The
formula _Doubling Time (hrs)_ = [_Culture Time (hrs)_ / [log2 (_Cell Number_ _end_ / _Cell number_ _start_)]] was used to quantify doubling time. SEAHORSE METABOLIC FLUX ASSAY Cells were
seeded in Seahorse XF96 Cell Culture Microplates at 36000 cells/well for HEK293 cells and 17000 cells/well for 143B cells 24 h prior to the experiment to achieve around 70–80% confluence on
the day of the experiment, and treatments (NR, FK866) were administered simultaneously in 100 μL of the culture media (described above). 4 wells were left unseeded in each experiment to
serve as background measurements. One hour prior to performing the experiment, cell culture growth media was replaced by unbuffered Seahorse XF DMEM media (pH 7.4) supplemented with 2 mM
glutamine, 1 mM sodium pyruvate, and 5 mM glucose and cell plates subsequently incubated in a low CO2 incubator for around 45 min to equilibrate pH. To evaluate mitochondrial oxygen
consumption rates (OCR) and extracellular acidification rates (ECAR), the XF Mito Stress Test was performed according to the manufacturer’s instructions on the Seahorse XF96 Analyzer. Basal
respiration was obtained by measuring at 3 consecutive time points prior to any pharmacological perturbation. OCR and ECAR was averaged across all three time points to give a single measure
for each well. MT_LB_NOX EXPERIMENTS Plasmids were designed and ordered from ATUM. mt_Lb_NOX23 and a red fluorescence reporter (CayenneRFP) were designed to be under the control of a CMV
promoter with a T2A site to allow them to be transcribed as one transcript and later separated during translation. Inclusion of a neomycin resistance gene enabled selection of transfected
cells. 143B osteosarcoma cells were plated and transfected at 30% confluence using TransIT-X2 after 24 h. Successfully transfected cells were selected for by addition of 400 µg/ml G418 for 2
weeks and continuous culture under 200 µg/ml G418. Clonal selection was not performed to generate a cell line with heterogeneous mt_Lb_NOX expression. CayenneRFP fluorescence was measured
at the same time as NADH FLIM, allowing correlations between mean NADH lifetime and CayenneRFP fluorescence (marker for mt_Lb_NOX expression) in each cell. CayenneRFP fluoresence in
individual cells was quantified with Fiji (ImageJ) by first marking a region of interest around each cell with the “Freehand Selection” tool and then measuring average pixel intensity.
Corresponding cells were analyzed in NADH FLIM images for mean lifetime as described in subsequent sections. The mean lifetime of each cell was normalized to the average τmean of all
untreated or antimycin A treated cells, respectively. NADH FLIM OF CELLS Cells were seeded in 35 mm dishes with glass bottom (Greiner, 627870) 48 h prior to the experiment in standard cell
culture medium (as above) supplemented with 25 mM HEPES. NR/FK866 treatments NR/FK866 treatments of varying concentrations (300 µM and 1 mM NR; 1 nM, 1.5 nM, 2 nM, and 5 nM FK866) were
administered in the same medium 24 h prior to the experiments. Directly before the experiment, media was removed and cells were imaged in Tyrodes buffer (135 mM NaCl, 5 mM KCl, 1.8 mM CaCl2,
20 mM Hepes, 5 mM glucose, 1 mM MgCl2, pH 7.4). Antimycin A treatments (5 µM), used to block respiration, were administered directly prior to measurements. Fluorescence lifetime imaging of
NADH was performed on a laser scanning microscope (Zeiss LSM 710) as described previously51. Briefly, NADH was excited using two-photon excitation with a pulsed (80 MHz, 100 fs pulse width)
titanium-sapphire laser at a power of <1.5 mW on the sample. Time-correlated single photon counting (TCSPC) with the hybrid detector HPM-100-40 was performed. The detector was coupled to
the NDD port of the LSM 710. FLIM images (512 × 512 pixel) were taken at a temporal resolution of 256 time channels. Final settings: 60 sec collection time; ≈ 15 μsec pixel dwell time; 135 ×
135 μm2 scanning area, Plan-Apochromat 63x/1.40 Oil DIC M27 lense, 730 nm excitation wavelength and 460/50 nm bandpass emission filter. Data were recorded using SPCM 9.8 and subsequently
analyzed using SPCImage 8.8 for all cytoplasmic data and SPCImage 8.0 for all other data assuming a biexponential decay. Standard analysis settings if not indicated otherwise (Figs. 1, 2, 3
and Fig. S1, S3): WLS fit method, biexponential decay with unfixed lifetime components (τ1 and τ2), square binning of 2, peak threshold adapted to background, shift fixed at a pixel of clear
NADH signal. The mean lifetime (τmean) was calculated and fitting of the calculated lifetime curve was confirmed by checking the mean χ2, which should be below 1.2. For subcellular
analysis, nuclei were selected by drawing regions of interest (ROIs) using the “Define mask” tool in areas clearly distinguishable as nuclei by eye. At least five nuclei were masked for
analysis per image. The mitochondria-rich regions were isolated from the nucleus and cytoplasm using the intensity threshold function because mitochondria tend to have higher fluorescence
than the surrounding cytoplasm and the nucleus. The cytoplasmic regions were selected for quantification by marking a region of interest using the “Define Mask” tool in areas clearly
distinguishable as lacking mitochondria by eye. Cytoplasmic regions from at least five cells were selected per image. 2D correlations in SPCImage 8.8 were used to isolate cytoplasmic regions
for visualization in representative images by limiting τmean to exclude the shorter nuclear lifetimes on one axis and limiting a1abs to exclude high intensity mitochondrial regions on the
second axis. For single cell analysis of nuclear, mitochondrial, and cytoplasmic τmean, the “Define Mask” tool was used to mark a region of interest around the selected cell or nucleus.
Metabolic Delta: Individual data points for metabolic delta were obtained by subtracting the τmean from each individual image for each treatment group (untr, NR, FK866) after antimycin A
treatment from the average τmean of each treatment group before antimycin A treatment. FLIM parameter analysis: Since mt_Lb_NOX and antimycin A treatments produced a continuous range of
alterations in τmean, unfixed lifetime components (τ1 and τ2) allowed direct correlation of the lifetime parameters, displayed for τ1 and a1/a2, with τmean. Pool-size induced lifetime
alterations were calculated as lifetime component xFK866/xNR-1 and subsequently normalized to respective alteration in τmean between the same images calculated as τmeanFK866/ τmeanNR-1.
Accordingly, the respiration-induced lifetime alteration was calculated as (xuntr-xAA-1)/(τmeanuntr/ τmeanAA-1) with x being any of the aforementioned individual lifetime components. NADH
FLIM OF LIVER Liver tissue was dissected from mice sacrificed by cervical dislocation and immediately placed in Tyrodes buffer. Tissue was then mounted on an imaging chamber flooded with
Tyrodes buffer and NADH FLIM was performed as described above but with a lower laser power <1.5 mW on the sample and using a 20x air lens (NA 0.8) with a 425.1 × 425.1 μm2 scanning area.
Due to high amounts of non-NADH autofluorescence in liver NADH FLIM images, the phasor plot was calculated in SPCImage. The phasor plot does not require an assumption of the number of
lifetime components or a fitting procedure but instead displays the lifetime of each pixel as a phasor within a half-circle52. Pixels directly on the half-circle are monoexponential, with
pixels to the right representing a shorter lifetime. Pixels within the half-circle are multiexponential. Since the phasor plot does not require a fitting procedure, it is ideal to
distinguish a mix of autofluorophores according to their lifetime characteristics. The region with NADH signal was identified by common NADH autofluorescence τ1 and τ2 values (the non-NADH
autofluorescence showed a very long τ1 ~ 1000 ps) and validated by comparing phasor plots of control tissue with phasor plots of antimycin A treated tissue. Antimycin A only impacts NADH
fluorescence, causing a shift in the location of the NADH signal region in the phasor plot. The pixels with an NADH autofluorescence signal profile were selected using a region of interest
(ROI) by checking “Select Cluster”. The photons of all pixels within the ROI were summed up and the biexponential decay was calculated with non-fixed lifetime components and non-fixed shift.
NAD+/NADH AND NADP+/NADPH QUANTIFICATION Quantification in cells: For biochemical quantification of NAD+, NADH and NADP+, NADPH using the NAD/NADH-GloTM Assay and the NADP/NADPH-GloTM Assay
(Promega), the provided protocol was followed. Briefly, cells were seeded 48 h prior and respective treatments were performed 24 h (NR, FK866) or 5 min (antimycin A) prior to the
experiment. The media was removed, cells were washed in ice-cold PBS and lysed in 100 µl base solution (0.2 N NaOH + 1% DTAB). Samples were split in half for measuring reduced or oxidized
nucleotides respectively. For the detection of NADH, samples were heated to 60 °C for 15 min and for the detection of NAD+ samples were first acidified by adding 25 µl of 0.4 N HCl and
subsequently heated to 60 °C for 15 min. Following neutralization using HCl-Trizma or Trizma base respectively, the assay kit was performed and luminescence recorded. For quantification in
subcellular fractions, mitochondrial isolation was performed by first washing cells in ice cold PBS and collecting using a cell scraper. Cell samples were then resuspended in 1 mL of ice
cold mitochondrial isolation buffer (75 mM sucrose, 225 mM mannitol, 10 mM MOPS, pH 7.4)53. EGTA was not included in the buffer to prevent interactions with the NAD or NADP cycling enzyme in
subsequent steps. All subsequent steps were performed on ice. Cells were lysed with 20 strokes of a pre-chilled glass Dounce homogenizer, lysate centrifuged at 1000 g, and resulting
supernatant centrifuged again at 1000 g to remove unbroken cells and nuclei. The mitochondrial fraction was obtained by centrifuging at high speed (10000 g). The supernatant after high-speed
centrifugation was collected as the cytosolic fraction, and the remaining mitochondrial pellet was washed in 1 mL mitochondrial isolation buffer and collected by spinning once more at 10000
g. Mitochondrial pellets were resuspended in 110 µl mitochondrial isolation buffer. Protein was quantified in mitochondrial and cytosolic fractions using the PierceTM BCA protein assay
(Thermo Scientific) according to manufacturer’s protocol for normalization. 50 µl of each cytosolic and mitochondrial sample were used to quantify NAD+, NADH and NADP+, NADPH using the
NAD/NADH-GloTM Assay and the NADP/NADPH-GloTM Assay (Promega) as described above, using 50 µl of base solution for lysis instead. For biochemical quantification of NAD+ and NADH in control
and mt_Lb_NOX cells using the NAD/NADH Quantification Kit (Sigma-Aldrich), the provided protocol was used. Cells were seeded 24 h prior to the experiment. On the day of the experiment, media
was removed and cells were then washed in PBS, trypsinized, and counted for 20,000 cells per sample. Each sample was lysed in 400 µl of provided extraction buffer. Half of the cell lysate
was separated and heated to 60 °C for 30 min to degrade NAD+. NAD(H) and NADH were then detected colorimetrically according the manufacturer’s protocol. Quantification in tissue: For
biochemical quantification of NAD+, NADH and NADP+, NADPH in liver the NAD/NADH Quantification Kit (Sigma-Aldrich) was used and an NAD(H) cycling assay was performed. Upon cervical
dislocation, pieces of the right liver lobe were dissected and flash frozen. The liver pieces were powdered on dry ice and the tissue powder was portioned into tubes and weighted. For the
quantification kit, liver powder was dissolved in the provided extraction buffer (200 µl/10 mg of tissue powder) and incubated on ice for 15 min. Afterwards the lysate was spun and the
supernatant split into to two tubes. One was used for NAD(P) + NAD(P)H detection and one was heated to 60 °C for 30 min to detect NADH only. Subsequently the NAD(P)(H) detection kits were
performed following the manufacturers protocol. For the cycling assay, two separate tubes were used. For NADH the powdered tissue was lysed in ice-cold 0.25 M KOH in 50% ethanol while for
NAD+ it was lysed in ice-cold 0.6 M Perchloric Acid. Samples were spun down for 15 min at 20,000 g at 4 °C, the supernatant transferred to new tubes and diluted 1:40 (1:25 for NADH) in ice
cold Na-Phosphate buffer (pH 8.0). Diluted extracts or standards were added to a cycling mixture (2% ethanol, 100 μg/mL alcohol dehydrogenase, 10 μg/mL diaphorase, 20 μM resazurin, 10 μM
flavin mononucleotide, 10 mM nicotinamide, 0.1% BSA in 100 mM phosphate buffer, pH 8.0) and fluorescence was quantified at 590 nm (ex:530 nm) in a 96 well plate. TARGETED LC/MS METABOLOMICS
Approximately 5 mg aliquots of dry liver powder were homogenized in 500 μl of 80% ice cold methanol in the Precellys homogenizer at 4 °C. Then, 100 μl of homogenates were extracted with 400
μl of ice-cold methanol, vortexed, centrifuged at 18,000 × _g_, and 400 μl aliquots of supernatants were dried under nitrogen at 45 °C in a 96-well plate. An Agilent PEEK poroshell HIILIC-z
column was used to separate nucleotides with gradient elution. Quantitation of metabolites in each assay module was achieved using multiple reaction monitoring of calibration solutions and
study samples on an Agilent 1290 Infinity UHPLC/6495 triple quadrupole mass spectrometer. Raw data were processed using Mass Hunter quantitative analysis software (Agilent). Calibration
curves (R2 = 0.99 or greater) were either fitted with a linear or a quadratic curve with a 1/_X_ or 1/_X_2 weighting. STATISTICS AND REPRODUCIBILITY Each point in graphs represents an
independent data point with the line indicating the mean and the error bars indicating the SEM. Data were tested for Gaussian distributions using the D’Agostino and Pearson omnibus normality
test. If all data passed the normality test (_p_ > 0.05), an unpaired two tailed t-test (two groups) or a One-Way ANOVA with Bonferroni correction for multiple testing was performed. If
any data did not pass the normality test, a Mann-Whitney test (two groups) or a Kruskal-Wallis test with Dunns’ correction for multiple testing was performed. To elucidate the effect of NR
in-vivo across mouse strains and age, 6- and 18-month-old mice of B6 and ANT1 mice were used. To reveal the effect of NR, each independent data point was normalized by dividing it by the
mean value for the respective saline-injected control group. A correlation between mean NADH lifetime and NAD+/NADH ratio was generated by correlating the averaged value for mean lifetime of
each treatment group (300 μM NR, 1 mM NR, untr, 1 nM FK855, 1.5 nM FK866, 2 nM FK866, and 5 nM FK866) with the averaged value for biochemically measured NAD+/NADH ratio. The correlation
between mean NADH lifetime and NAD(H) levels was similarly calculated. Significances between treatment groups (*), between antimycin A treated and untreated groups (#), or between
mitochondrial and cytosolic fractions (#) are displayed as n.s. _P_ > 0.05, ∗/# _p_ < 0.05, ∗∗/## _p_ < 0.01, ∗∗∗/### _p_ < 0.001. To ensure reproducibility, NADH FLIM imaging of
untreated (HEK293, 143B, and mtLbNOX) and corresponding treated (NR, FK866, AA) cells was repeated in five independent experiments on separate days, and five images taken from different
areas of each treatment well during each experiment. Quantification of NAD(P)(H) was performed using several methods, including by two types of commercially available biochemical kits and by
cycling assay, to ensure data reproducibility. Sample sizes for all experiments were determined based on similar experiments in the field. Sample size was maximized where possible and in
accordance with experimental bandwidth. Technical replicates were averaged and biological replicates were treated independently. A minimum of three biological replicates were used for all
quantifications, and a minimum of 3 independent experiments were performed for all respirometry data. DATA AVAILABILITY The authors declare that all data supporting the findings of this
study are included in the paper, its Supplementary Information, and its Supplementary Data. CODE AVAILABILITY The authors declare that no new code was created or used to analyze the data.
REFERENCES * Wallace, D. C. A mitochondrial bioenergetic etiology of disease. _J. Clin. Invest_ 123, 1405–1412 (2013). Article CAS PubMed PubMed Central Google Scholar * Katsyuba, E. et
al. NAD. _Nat. Metab._ 2, 9–31 (2020). Article CAS PubMed Google Scholar * Kim, J. & DeBerardinis, R. J. Mechanisms and implications of metabolic heterogeneity in cancer. _Cell
Metab._ 30, 434–446 (2019). Article CAS PubMed PubMed Central Google Scholar * Niederschweiberer, M. A. et al. NADH fluorescence lifetime imaging microscopy reveals selective
mitochondrial dysfunction in neurons overexpressing alzheimer’s disease-related proteins. _Front Mol. Biosci._ 8, 671274 (2021). Article CAS PubMed PubMed Central Google Scholar *
Chance, B. Spectrophotometry of intracellular respiratory pigments. _Science_ 120, 767–775 (1954). Article CAS PubMed Google Scholar * Mayevsky, A. & Chance, B. Oxidation-reduction
states of NADH in vivo: from animals to clinical use. _Mitochondrion_ 7, 330–339 (2007). Article CAS PubMed Google Scholar * Chance, B. et al. Oxidation-reduction ratio studies of
mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. _J. Biol. Chem._ 254, 4764–4771 (1979). Article CAS PubMed Google Scholar * Vergen, J. et al.
Metabolic imaging using two-photon excited NADH intensity and fluorescence lifetime imaging. _Microsc Microanal._ 18, 761–770 (2012). Article CAS PubMed Google Scholar * Lakowicz, J. R.
et al. Fluorescence lifetime imaging of free and protein-bound NADH. _Proc. Natl. Acad. Sci. USA_ 89, 1271–1275 (1992). Article CAS PubMed PubMed Central Google Scholar * Schaefer, P.
M. et al. Mitochondrial matrix pH as a decisive factor in neurometabolic imaging. _Neurophotonics_ 4, 045004 (2017). Article PubMed PubMed Central Google Scholar * Drozdowicz-Tomsia, K.
et al. Multiphoton fluorescence lifetime imaging microscopy reveals free-to-bound NADH ratio changes associated with metabolic inhibition. _J. Biomed. Opt._ 19, 086016 (2014). Article
PubMed Google Scholar * Sanchez, W. Y. et al. Analysis of the metabolic deterioration of ex vivo skin from ischemic necrosis through the imaging of intracellular NAD(P)H by multiphoton
tomography and fluorescence lifetime imaging microscopy. _J. Biomed. Opt._ 15, 046008 (2010). Article PubMed Google Scholar * Schaefer, P. M. et al. NADH autofluorescence-A marker on its
way to boost bioenergetic research. _Cytometry A._ 95, 34–46 (2018). * Sharick, J. T. et al. Protein-bound NAD(P)H lifetime is sensitive to multiple fates of glucose carbon. _Sci. Rep._ 8,
5456 (2018). Article PubMed PubMed Central Google Scholar * Blacker, T. S. et al. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. _Nat. Commun._ 5, 3936
(2014). Article CAS PubMed Google Scholar * Hong, S. M. et al. Increased nicotinamide adenine dinucleotide pool promotes colon cancer progression by suppressing reactive oxygen species
level. _Cancer Sci._ 110, 629–638 (2019). Article CAS PubMed Google Scholar * Pirinen, E. et al. Niacin cures systemic NAD+ deficiency and improves muscle performance in adult-onset
mitochondrial myopathy. _Cell Metab._ 31, 1078–1090.e5 (2020). Article CAS PubMed Google Scholar * Imai, S. & Guarente, L. NAD+ and sirtuins in aging and disease. _Trends Cell Biol._
24, 464–471 (2014). Article CAS PubMed PubMed Central Google Scholar * McReynolds, M. R., Chellappa, K. & Baur, J. A. Age-related NAD. _Exp. Gerontol._ 134, 110888 (2020). Article
CAS PubMed PubMed Central Google Scholar * Yoshino, J., Baur, J. A. & Imai, S. I. NAD+ intermediates: The biology and therapeutic potential of NMN and NR. _Cell Metab._ 27, 513–528
(2018). Article CAS PubMed Google Scholar * Covarrubias, A. J. et al. NAD+ metabolism and its roles in cellular processes during ageing. _Nat. Rev. Mol. Cell Biol._ 22, 119–141 (2021).
Article CAS PubMed Google Scholar * Goodman, R. P., Calvo, S. E. & Mootha, V. K. Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. _J. Biol. Chem._ 293,
7508–7516 (2018). Article CAS PubMed PubMed Central Google Scholar * Titov, D. V. et al. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH
ratio. _Science_ 352, 231–235 (2016). Article CAS PubMed PubMed Central Google Scholar * Christensen, C. E. et al. Non-invasive in-cell determination of free cytosolic [NAD+]/[NADH]
ratios using hyperpolarized glucose show large variations in metabolic phenotypes. _J. Biol. Chem._ 289, 2344–2352 (2014). Article CAS PubMed Google Scholar * Luongo, T. S. et al.
SLC25A51 is a mammalian mitochondrial NAD. _Nature_ 588, 174–179 (2020). Article CAS PubMed PubMed Central Google Scholar * Becker, W. _The bh TCSPC Handbook_. Becker and Hickl GmbH.
(2015) * Blacker, T. S. & Duchen, M. R. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. _Free Radic. Biol. Med_ 100, 53–65 (2016). Article CAS PubMed
PubMed Central Google Scholar * Veskoukis, A. S. et al. Spectrophotometric assays for measuring redox biomarkers in blood and tissues: the NADPH network. _Redox Rep._ 23, 47–56 (2018).
Article CAS PubMed Google Scholar * Zhao, Y. et al. SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. _Cell Metab._ 21,
777–789 (2015). Article CAS PubMed PubMed Central Google Scholar * Tao, R. Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. _Nat. Methods_ 14,
720–728 (2017). Article CAS PubMed PubMed Central Google Scholar * König, K. Review: Clinical in vivo multiphoton FLIM tomography. _Methods Appl Fluoresc._ 8, 034002 (2020). Article
PubMed Google Scholar * Katsyuba, E. et al. NAD homeostasis in health and disease. _Nat. Metab._ 2, 9–31 (2020). Article CAS PubMed Google Scholar * Pastore, M. N. et al. Non-invasive
metabolic imaging of melanoma progression. _Exp. Dermatol_ 26, 607–614 (2017). Article CAS PubMed Google Scholar * Yaseen, M. A. et al. Fluorescence lifetime microscopy of NADH
distinguishes alterations in cerebral metabolism in vivo. _Biomed. Opt. Express_ 8, 2368–2385 (2017). Article CAS PubMed PubMed Central Google Scholar * Lautrup, S. et al. NAD+ in brain
aging and neurodegenerative disorders. _Cell Metab._ 30, 630–655 (2019). Article CAS PubMed PubMed Central Google Scholar * Heikal, A. A. Intracellular coenzymes as natural biomarkers
for metabolic activities and mitochondrial anomalies. _Biomark. Med._ 4, 241–263 (2010). Article CAS PubMed Google Scholar * Mattson, M. P. & Arumugam, T. V. Hallmarks of brain
aging: Adaptive and pathological modification by metabolic states. _Cell Metab._ 27, 1176–1199 (2018). Article CAS PubMed PubMed Central Google Scholar * Dong, Y., Digman, M. A. &
Brewer, G. J. Age- and AD-related redox state of NADH in subcellular compartments by fluorescence lifetime imaging microscopy. _Geroscience_ 41, 51–67 (2019). Article CAS PubMed PubMed
Central Google Scholar * Sanchez, T. et al. Metabolic imaging with the use of fluorescence lifetime imaging microscopy (FLIM) accurately detects mitochondrial dysfunction in mouse oocytes.
_Fertil. Steril._ 110, 1387–1397 (2018). Article PubMed PubMed Central Google Scholar * Sun, N., Youle, R. J. & Finkel, T. The mitochondrial basis of aging. _Mol. Cell_ 61, 654–666
(2016). Article CAS PubMed PubMed Central Google Scholar * Niesner, R. et al. Noniterative biexponential fluorescence lifetime imaging in the investigation of cellular metabolism by
means of NAD(P)H autofluorescence. _Chemphyschem_ 5, 1141–1149 (2004). Article CAS PubMed Google Scholar * Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. _Science_ 350,
1208–1213 (2015). Article CAS PubMed Google Scholar * Hu, Q. et al. Genetically encoded biosensors for evaluating NAD+/NADH ratio in cytosolic and mitochondrial compartments. _Cell Rep.
Methods_ 1, 100116 (2021). Article CAS PubMed PubMed Central Google Scholar * Wright, B. K. et al. Phasor-FLIM analysis of NADH distribution and localization in the nucleus of live
progenitor myoblast cells. _Microsc Res. Tech._ 75, 1717–1722 (2012). Article CAS PubMed PubMed Central Google Scholar * VanLinden, M. R. et al. Subcellular distribution of NAD+ between
cytosol and mitochondria determines the metabolic profile of human cells. _J. Biol. Chem._ 290, 27644–27659 (2015). Article CAS PubMed PubMed Central Google Scholar * Anderson, K. A.
et al. Metabolic control by sirtuins and other enzymes that sense NAD(+), NADH, or their ratio. _Biochim. Biophys. Acta_ 1858, 991–998 (2017). Article CAS PubMed Central Google Scholar *
Chacko, J. V. & Eliceiri K. W., Autofluorescence lifetime imaging of cellular metabolism: Sensitivity toward cell density, pH, intracellular, and intercellular heterogeneity. _Cytometry
A._ 95, 56–69 (2018). * Yang, X., Ha, G. & Needleman, D. J. A coarse-grained NADH redox model enables inference of subcellular metabolic fluxes from fluorescence lifetime imaging.
_Elife_ 10, e73808 (2021). Article PubMed PubMed Central Google Scholar * Graham, B. H. et al. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in
the heart/muscle isoform of the adenine nucleotide translocator. _Nat. Genet_ 16, 226–234 (1997). Article CAS PubMed Google Scholar * Schaefer, P. M. et al. Metabolic characterization of
intact cells reveals intracellular amyloid beta but not its precursor protein to reduce mitochondrial respiration. _PLoS One_ 11, E0168157 (2016). Article PubMed PubMed Central Google
Scholar * Schaefer, P. M. et al. Nicotinamide riboside alleviates exercise intolerance in ANT1-deficient mice. _Mol. Metab._ 64, 101560 (2022). Article CAS PubMed PubMed Central Google
Scholar * Digman, M. A. et al. The phasor approach to fluorescence lifetime imaging analysis. _Biophys. J._ 94, L14–L16 (2008). Article CAS PubMed Google Scholar * Lin, C. S. et al.
Mouse mtDNA mutant model of Leber hereditary optic neuropathy. _Proc. Natl. Acad. Sci. USA_ 109, 20065–20070 (2012). Article CAS PubMed PubMed Central Google Scholar Download references
ACKNOWLEDGEMENTS We would like to thank Christopher Petucci and the Metabolomics Core in the Cardiovascular Institute at the University of Pennsylvania for performing and analyzing the
global and targeted metabolomics experiments. The authors would like to express their gratitude to Niagen® for providing the NR. Lastly, the authors would like to thank Piotr Kopinski for
his assistance in creating the mt_Lb_NOX plasmids. This work was supported by the German Research Foundation (SCHA 2182/1-1) and the Foerderer Award 2020 awarded to PM Schaefer, the National
Institutes of Health grants NS021328, MH108592, OD010944 and U.S. Department of Defense grants W81XWH-16-1-0401 and W81XWH-21-1-0128 (PR202887.e002) awarded to DC Wallace. AUTHOR
INFORMATION AUTHORS AND AFFILIATIONS * Center for Mitochondrial and Epigenomic Medicine, Department of Pathology and Laboratory Medicine, Children’s Hospital of Philadelphia, Philadelphia,
PA, USA Angela Song, Nicole Zhao, Douglas C. Wallace & Patrick M. Schaefer * Department of Bioengineering, University of Pennsylvania, Philadelphia, PA, USA Angela Song * Department of
Biological Sciences, Dartmouth College, Hanover, NH, USA Diana C. Hilpert * Department of Physiology and Institute for Diabetes, Obesity, and Metabolism, Perelman School of Medicine,
University of Pennsylvania, Philadelphia, PA, USA Caroline Perry & Joseph A. Baur * Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA,
USA Douglas C. Wallace Authors * Angela Song View author publications You can also search for this author inPubMed Google Scholar * Nicole Zhao View author publications You can also search
for this author inPubMed Google Scholar * Diana C. Hilpert View author publications You can also search for this author inPubMed Google Scholar * Caroline Perry View author publications You
can also search for this author inPubMed Google Scholar * Joseph A. Baur View author publications You can also search for this author inPubMed Google Scholar * Douglas C. Wallace View author
publications You can also search for this author inPubMed Google Scholar * Patrick M. Schaefer View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS P.M.S. conceptualized the project and designed research; A.S., N.Z, C.P. and P.M.S. performed the experiments; A.S., D.C.H. and P.M.S. analyzed data; A.S., J.A.B., D.C.W., and
P.M.S. interpreted the data, A.S., D.C.W., and P.M.S. wrote the paper. All authors read, edited and approved the manuscript. CORRESPONDING AUTHORS Correspondence to Douglas C. Wallace or
Patrick M. Schaefer. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare the following competing interests: J.A.B. is consultant to Pfizer and Cytokinetics, an inventor on a patent
for using NAD+ precursors in liver injury and has received research funding and materials from Elysium Health and Metro International Biotech, both of which have an interest in NAD+
precursors. D.C.W. is part of the scientific advisory boards for Pano Therapeutics and Medical Excellence Capital. All other authors declare no competing interests. PEER REVIEW PEER REVIEW
INFORMATION _Communications Biology_ thanks Xingbo Yang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Gene Chong and
Christina Karlsson Rosenthal. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps
and institutional affiliations. SUPPLEMENTARY INFORMATION PEER REVIEW FILE SUPPLEMENTARY INFORMATION DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA RIGHTS AND PERMISSIONS
OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or
other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Song, A., Zhao, N.,
Hilpert, D.C. _et al._ Visualizing subcellular changes in the NAD(H) pool size versus redox state using fluorescence lifetime imaging microscopy of NADH. _Commun Biol_ 7, 428 (2024).
https://doi.org/10.1038/s42003-024-06123-7 Download citation * Received: 28 February 2023 * Accepted: 29 March 2024 * Published: 09 April 2024 * DOI:
https://doi.org/10.1038/s42003-024-06123-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Cristiano ronaldo: juventus star sent off on champions league debutRonaldo signed for Juventus from Real Madrid in July for £105million, with the Serie A giants hopeful he can propel them...
Neobank n26 gives customers in france local iban numbersTHE GERMAN BANK HOPES MONEY TRANSFERS WILL BECOME EASIER - BUT IT WILL CHANGE HOW CUSTOMERS DECLARE THEIR ACCOUNT ON TAX...
Chris packham blasts the chase stars for swimming with dolphinsChris said: “I've met the Chasers, they're a really nice bunch of people, but they're not natural histori...
Mansoor, rush debate without daigle - newport beach newsA clear divergence of beliefs piled up between Assemblyman Allan Mansoor, a Republican from Costa Mesa, and political ne...
Javascript support required...
Latests News
Visualizing subcellular changes in the nad(h) pool size versus redox state using fluorescence lifetime imaging microscopy of nadhABSTRACT NADH autofluorescence imaging is a promising approach for visualizing energy metabolism at the single-cell leve...
Next's £34 patent loafers that are 'comfy and stylish for everyday wear'SHOPPERS SAY THE LOAFERS LOOK 'POLISHED AND PROFESSIONAL' 12:15, 21 May 2025 This article contains affiliate l...
Attention Required! | CloudflarePlease enable cookies. Sorry, you have been blocked You are unable to access defatoonline.com.br Why have I been blocked...
How saving an endangered species can mitigate climate changeAwareness programmes have been organised in many coastal villages, such as Kazhumanguda, Karanguda, Mallipattinam, Chinn...
How California’s income tax and sales tax rates compareThis article was originally on a blog post platform and may be missing photos, graphics or links. See About archive blog...