Novel computational and drug design strategies for inhibition of human papillomavirus-associated cervical cancer and dna polymerase theta receptor by apigenin derivatives
Novel computational and drug design strategies for inhibition of human papillomavirus-associated cervical cancer and dna polymerase theta receptor by apigenin derivatives"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The present study deals with the advanced in-silico analyses of several Apigenin derivatives to explore human papillomavirus-associated cervical cancer and DNA polymerase theta
inhibitor properties by molecular docking, molecular dynamics, QSAR, drug-likeness, PCA, a dynamic cross-correlation matrix and quantum calculation properties. The initial literature study
revealed the potent antimicrobial and anticancer properties of Apigenin, prompting the selection of its potential derivatives to investigate their abilities as inhibitors of human
papillomavirus-associated cervical cancer and DNA polymerase theta. In silico molecular docking was employed to streamline the findings, revealing promising energy-binding interactions
between all Apigenin derivatives and the targeted proteins. Notably, Apigenin 4′-O-Rhamnoside and Apigenin-4′-Alpha-l-Rhamnoside demonstrated higher potency against the HPV45 oncoprotein E7
(PDB ID 2EWL), while Apigenin and Apigenin 5-O-Beta-d-Glucopyranoside exhibited significant binding energy against the L1 protein in humans. Similarly, a binding affinity range of − 7.5
kcal/mol to − 8.8 kcal/mol was achieved against DNA polymerase theta, indicating the potential of Apigenin derivatives to inhibit this enzyme (PDB ID 8E23). This finding was further
validated through molecular dynamic simulation for 100 ns, analyzing parameters such as RMSD, RMSF, SASA, H-bond, and RoG profiles. The results demonstrated the stability of the selected
compounds during the simulation. After passing the stability testing, the compounds underwent screening for ADMET, pharmacokinetics, and drug-likeness properties, fulfilling all the
necessary criteria. QSAR, PCA, dynamic cross-correlation matrix, and quantum calculations were conducted, yielding satisfactory outcomes. Since this study utilized in silico computational
approaches and obtained outstanding results, further validation is crucial. Therefore, additional wet-lab experiments should be conducted under in vivo and in vitro conditions to confirm the
findings. SIMILAR CONTENT BEING VIEWED BY OTHERS COMPUTATIONAL ANALYSIS OF LUPENONE DERIVATIVES AS POTENTIAL INHIBITOR OF HUMAN PAPILLOMAVIRUS ONCOPROTEIN E6 ASSOCIATED CERVICAL CANCER
Article Open access 02 May 2025 IN-SILICO STUDY UNVEILS POTENTIAL PHYTOCOMPOUNDS IN _ANDROGRAPHIS PANICULATA_ AGAINST E6 PROTEIN OF THE HIGH-RISK HPV-16 SUBTYPE FOR CERVICAL CANCER THERAPY
Article Open access 26 July 2024 MULTITARGETED DOCKING APPROACH REVEALS DROXIDOPA AGAINST DNA REPLICATION AND REPAIR-RELATED PROTEIN OF CERVICAL CANCER Article Open access 16 October 2024
INTRODUCTION Cervical cancer contributed to 604,127 cases (6.5% of all cancer cases) each year. It inflicted 341,831 deaths (7.7% of all cancer deaths) in 2020 globally, placing it as the
fourth most lethal type of cancer in the female population1. The worldwide cancer burden is quickly growing as a consequence of continuing demographic and epidemiological shifts, and it is
anticipated that this will contribute to a large percentage (> 4,74,000) of deaths among women by the year 20302. Cervical cancer is primarily spread between affluent and less affluent
nations, as evidenced by this worldwide cancer burden. evidence shows that viral infections are responsible for 15–20% of all human malignancies. Different phases of cancer development may
be accelerated by infection with oncogenic viruses. There are many other forms of Human Papilloma Virus (HPV), but around 15 have been related to cancer. Even if screening procedures are
very successful, cervical cancer is still a significant public health issue3. HPV is a small, non-enveloped, icosahedral, double-stranded DNA virus that may transmit through sexual
activities. It infects different parts of the body’s organs, such as squamous epithelia, including the skin and upper respiratory and anogenital tract mucous membranes. About 100 various
other forms of HPV and around 40 of them are known to infect the anogenital region4. Besides, it has been associated with several different malignancies, the most significant of which is
cervical cancer. It is also reported that infection with HPV is the root cause of almost all cases of cervical cancer. The occurrence of cervical cancer has been linked to 18 different kinds
of human papillomavirus, with types HPV-16 and HPV-18, in particular, being considered high-risk/oncogenic. Low-risk/non-oncogenic HPV strains cause genital warts, especially types HPV-6
and HPV-11. Within a year to two years after infection, cell-mediated immunity typically clears or suppresses most cervical HPV infections5. The HPV may often be confirmed in clinical
specimens using hybrid capture or polymerase chain reaction of HPV genomes6. HPV genotyping, on the other hand, is accomplished through hybridization with type-specific oligonucleotide
probes. All probe assays rely on identifying target nucleic acid patterns by complementing probe nucleic acid patterns that may be replicated through PCR7. This identification can be
accomplished in a variety of ways. The target DNA is subjected to several cycles of denaturation, primer hybridization, and primer extension in the PCR, which results in the specific to an
individual of the target DNA. After 30 cycles of PCR, a proportion of target DNA equal to or more than one million duplicates is created. After that, either a standard dot blot or a Southern
blot is used to determine the results of the amplified DNA. One sort of test that uses a modulation scheme to identify DNA or RNA substrates is called a hybrid capture technique8. The
Hybrid capture method employs a hybridization solution consisting of RNA tags with DNA targets for HPV identification. This is complemented by an immunologically oriented back-end test
comparable to an ELISA. Regarding the detection of HPV, the PCR approach has a better analytic sensitivity than the hybrid capture approach; nevertheless, the hybrid capture method may be
more efficient in determining women who have concomitant squamous epithelial tumors9. Secondly, a particular kind of genetic recombination known as homologous recombination (HR) occurs when
two identical or comparable double-stranded or single-stranded nucleic acid molecules exchange genetic information. The biological system accurately repairs DNA breaks (both strands) through
HR where a specific process known as homologous recombinational repair (HRR) contributes10. HR deficiency has emerged as a crucial biomarker for multiple types of cancers such as ovarian,
breast, pancreatic, and prostate11. Mutation in two particular genes, BRCA1 and BRCA2 (combinedly addressed as BRCA1/2), is responsible for HR-mediated DNA repair deficiency leading to the
development and initiation of several types of tumors12. Interestingly, Poly (ADP-ribose) polymerase inhibitors (PARPi) have shown excellent sensitivity against BRCA1/2-mutated tumors, and
many PARPi has been approved in recent decades for clinical use13,14. But the main challenge to these medications’ clinical success in patients with HR-deficient tumors is the rapid
development of resistance. Therefore, across numerous therapeutic situations, including primary chemotherapy, neoadjuvant treatment, and combination therapy with immunotherapies, the
effectiveness of PARPi is now being assessed15,16,17,18. Recent research has suggested the synthetic lethality of HR deficiency with DNA polymerase theta making an emerging novel drug target
for treating HR-deficient tumors19. The DNA polymerase theta contains three domains (N-terminus containing a helicase-like ATPase domain, central domain, and C terminus containing a
nuclease domain) and a nuclease domain). It differs from other polymerases in structure and function as it suppresses mitotic crossovers for preserving genomic integrity20,21,22. Any
patients suffering from breast and ovarian tumors with HR deficiency have high expression of DNA polymerase theta, which is a backup HR mediator in the DNA double-strand break repair
process23. As DNA polymerase theta is synthetic and lethal with HR, inhibition of DNA polymerase theta in patients with defective HR can induce tumor cell death23,24. Moreover, the DNA
polymerase theta inhibition approach harmonizes with PARPi activity in HR-deficient tumor elimination23,24. Synthetic lethality among HR deficiency and DNA polymerase theta inhibition
depends on several mechanisms through which DNA polymerase theta operates to keep the genome stable and stop tumorigenesis21. Besides, the DNA polymerase theta is critical in mutagenic
microhomology-mediated end-joining (MMEJ), an significant DNA double-strand break-repairing mechanism25. However, the mechanism by which the DNA polymerase theta maintains synthetic
lethality with HR-deficient tumors remains unclear, but this evidence suggests that the versatile functionality of the DNA polymerase theta is vital for HR-deficient tumor survival. However,
The DNA polymerase theta demonstrates distinct characteristics of drug ability, offering a compelling case for creating DNA polymerase theta inhibitors26. Therefore, developing an inhibitor
targeting DNA polymerase theta should be a rational option for curing HR-deficient tumor cells. There is currently no therapy available for HPV27. So, potential treatment or drug is highly
needed to manage HPV and its related cancer. But, developing an effective medication with a high degree of potentiality is a very time-consuming matter, and required huge research funds.
Besides, during the developing phases, many drugs fail, and can’t go final stages due to unwanted effects28. Resulting, the research community may lose huge amounts of resources and costs.
But, in the modern era of drug development, this huge cost could be minimized by early investigation of physiochemical, and toxicity prediction29. So, in this investigation, the most popular
in silico application, and investigation are applied and determined the drug-like properties of Apigenin derivatives for the treatment of human papillomavirus, and its associated cancer.
Here, we also performed a comprehensive computational investigation supported by a rigorous literature-based approach aiming to identify the most potent DNA polymerase theta inhibitor from
selected Apigenin derivatives. These Apigenin derivatives were evaluated based on their ability to inhibit DNA polymerase theta ATPase activity and prevent the MMEJ repairing mechanism which
will induce HR-deficient tumor cell death. LITERATURE STUDIES AND LIGAND-RECEPTOR SELECTION CRITERIA GENOMIC STRUCTURE OF HPV The papillomavirus genome comprises three distinct sections and
is formed of a tiny, double-stranded, highly conserved DNA that is around 8000 base pairs in length. Understanding the molecular biology of this small DNA molecule is complicated. There are
seven proteins totaling 4000 base pairs (bp) that are involved in transcription and replication and cell metamorphosis; they include six early proteins, three regulatory proteins (E1, E2,
and E4), and three oncoproteins (E5, E6, and E7). The viral capsid is composed of two proteins, L1 and L2, encoded by a separate 3,000 bp section of DNA. A 1000 bp section called the long
control region (LCR) encodes the viral DNA replication and transcriptional regulatory components. The L1 protein is necessary for the development of viral pathogenicity, and it is
responsible for the promotion of virion attachment to heparin sulfate receptors in the basal membrane30. Again, the E1 protein forms a hexameric complex that attracts topoisomerase I, DNA
polymerase, and replication protein A (RPA), all of which are required for viral replication31. The E1 protein also urges DNA breaks in host chromatin, which aids viral integration, and the
E2 transcription factor regulates the E6 and E7 ORFs. When abundant, E2 binds to the 5′-ACCG(N)4CGGT-3′ palindromic sequence found in E2 binding sites (E2BS) in LCR, including the P97
promoter32. The E4 protein, the most expressed viral protein, is described in suprabasal and granular epithelial layers. The E4 interacts with keratin-associated amyloid fibers, causing cell
fragility and contributing to virion release33. DEVELOPMENT OF HPV IN CERVICAL CANCER The process of cervical cancerogenesis, in which HPV gene integration occurs between other cellular
alterations and epigenetic factors, is a complicated mechanism of unregulated cellular proliferation. Mutations in the DNA caused by the cell’s environment and HPV infection may allow the
virus' DNA to integrate with the host’s DNA synthesis machinery and cause replication of the virus. Thus, the virus can bypass cellular and immunological defenses while encouraging cell
growth and blocking apoptosis3. The Two primary oncogenic protein products E6 and E7 control the cell cycle by maintaining the process of normal apoptosis and they play an essential role in
promoting oncogenesis in cells. By duplicating their genetic material (DNA), viruses may produce cells to exhibit characteristics of cancer, including uncontrol growth, angiogenesis,
invasion, metastasis, and resistance to apoptosis and growth suppressors34. ROLE OF DNA POLYMERASE THETA IN DISEASE DEVELOPMENT DNA polymerase theta is a family of DNA polymerases (Pol θ)
that has been essential to maintaining DNA repair and damage tolerance. When any problematic condition occurs in the double strand of DNA, it helps to repair it. Some studies have reported
that when the DNA polymerase theta is overexpressed in cancer cells, it may promote the resistance of chemotherapeutic or cancerous agents. As a result, it makes it difficult to treat
cancer. So, the DNA polymerase theta might be a potential target receptor for the discovery of a drug to treat breast and ovarian cancers, etc.35,36. APIGENIN PHARMACOLOGICAL EVIDENCE
Apigenin is a flavonoid that may be found in a wide variety of fruits, vegetables, and plants used in traditional Chinese Medicine. This multifunctional molecule has several different
biological functions, including anti-inflammatory, antioxidant, antibacterial, and antiviral properties. As a result, Apigenin has a long history of usage in the context of alternative and
conventional medical practices. Apigenin has been connected to having an antitumor effect against a broad spectrum of cancers. This effect is thought to be achieved through apoptosis and
autophagy stimulation, cell cycle arrest, inhibition of cell migration and invasion, and an increase in immunological response37,38, and it may demonstrated to be capable of preventing,
inhibiting, or reversing the effects of chemically induced genotoxicity in vitro cell models, in vivo investigations, and AMES tests employing bacterial models. This anti-mutagenic effect
has been demonstrated and proven. The anti-carcinogenic function of Apigenin has received a lot of attention recently and has been discovered to be protective against different kinds of
cancer, including breast cancer, cervical cancer, prostate cancer, skin cancer, thyroid cancer, colon cancer, leukemia, lung cancer, endometrial cancer, neuroblastoma, and adrenocortical
cancer39,40. As Apigenin is composed of a multifunctional role against a number of diseases. So, this compound is chosen in our current computational experiment as a target biomolecule.
METHOD AND MATERIAL LIGAND PREPARATION AND MOLECULAR OPTIMIZATION The technique of optimization was worked out to achieve the best possible outcomes with regard to the performance of the
molecular docking approach as well as the arrangement of molecules in a three-dimensional framework. In the outset, a three-dimensional framework of Apigenin analogs was retrieved by
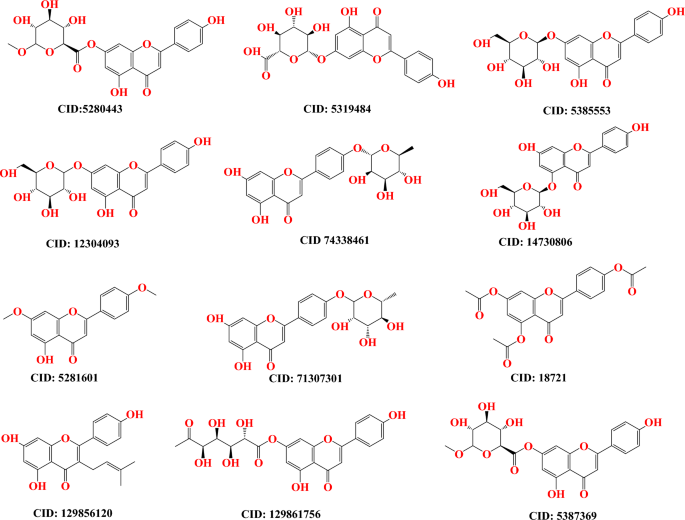
obtaining from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) (Fig. 1)41. This allowed for the structure to be observed in three dimensions. Prior to molecular docking, the ligands
were optimized using the Gaussian09 software with the DFT/B3LYP-6311G method. However, no aqueous operation optimization was conducted as it is not necessary for ligand optimization. The
purpose of these optimizations was to prepare the ligands for molecular docking. The molecular docking method was employed to select poses with the highest binding affinity (measured in
kcal/mol) between apigenin and apigenin 5-O-Beta-D Glucopyranoside ligands and the L1 protein of human papillomavirus (PDB ID 6L31). Subsequently, the best-selected poses underwent molecular
dynamics simulation. No separate ligand optimization was performed for the ligands in both complex structures. The topology files of the complex structures were prepared using Charmm-gui,
eliminating the need for a separate optimization for the ligands. PROTEIN PREPARATION AND MOLECULAR DOCKING STUDY The primary objective of the molecular docking technique is to make an
informed prediction regarding the composition of the ligand-receptor complex through the utilization of computational approaches. The docking process encompasses two distinct yet
interdependent steps. Firstly, it involves sampling several conformations that the ligand can adopt when bound to the protein’s active site. Secondly, it entails the classification of these
residues based on a performance index. By executing these intricately woven steps, molecular docking sheds between ligands and receptors are performed, and unraveling the prediction of their
interactions and aiding drug discovery and design endeavors42. The PyRx application was employed to accomplish molecular docking43. Before that, the crystal structure of HPV45 oncoprotein
E7 (PDB ID 2EWL), the L1 protein of human papillomavirus (PDB ID 6L31), and DNA polymerase theta (PDB ID 8E23) were collected from the RCSB protein data bank36,44,45. All of the crystal
solvent constituents were removed, together with the native agonist and any extra compounds from the crystal structure, and prepared for docking study (Fig. 2 showing the targeted
structure). During molecular docking studies, the grid box coordinates were strategically set to cover both the entire protein and the site of interest, ensuring accurate ligand placement.
The grid center points were set to X = − 34.66, Y = − 18.07, Z = 23.3766, and the box dimensions (Å) X = 26.45, Y = 53.3637, Z = 39.77 for (PDB-ID: 2EWL), the grid center for (PDB ID 6L31);
X = − 11.279, Y = − 28.1119, Z = − 28.2088, and box dimension X = 78.2212, Y = 22.88 and Z = 87.982 and the grid center for (PDB ID 8E23); X = − 19.552, Y = − 26.86772, Z = − 34.998, and box
dimension X = 80.99, Y = 27.99 and Z = 83.112 were set so that the grid box could wrap the whole substrate binding pocket of the protein structure. DETERMINATION OF ADMET, AND
PHARMACOKINETICS, AND DRUG-LIKENESS During the stage of drug discovery and development, it is completely obvious that the pharmacokinetic (PK) characteristics of potential therapies, and
more specifically ADMET (absorption, distribution, metabolism, excretion, and toxicity), are critical factors to consider46. Consequently, the Apigenin derivatives that were being explored
were initially placed through a critical drug-likeness screening using the SwissADME and pkCSM webservers47. This testing was based on calculated physicochemical and ADMET-related
parameters. The SMILES strings of the ligands that were utilized as the input for the molecular markers for both websites. In point of fact, unfavorable pharmacokinetic features of potential
drug candidates are a significant contributor to the rate of failure throughout clinical trials48. Because of this, it is vital to conduct pre-clinical evaluations of the pharmacokinetic
characteristics and drug-likeness of proposed medications. MOLECULAR DYNAMICS SIMULATION (MDS) PROTOCOL To understand the behavioral changes that occur in the protein–ligand complex when it
is exposed to a dynamic environment on an atomic level, MD is a good computer simulation method currently receiving a lot of attention in drug development research49. It is also an
indispensable instrument for determining the intra- or interatomic interaction stability of the protein–ligand complex over a user-specified period50. The best-scoring docking models of the
most potential Apigenin, and Apigenin-5-O-beta-D-glucopyranoside, apo form L1 protein of human papillomavirus (PDB ID 6L31), were chosen as the starting points for a 100-ns all-atom
molecular dynamics simulation using the GROMACS-2019 software (GNU, General Public License; http://www.gromacs.org). The CHARMM36 force field and the Gromacs version 2019 software package
were employed to conduct simulations running for 100 ns within a periodic water box51,52. The force field for Apigenin and Apigenin-5-O-beta-D-glucopyranoside, as well as the apo form L1
protein of human papillomavirus, was generated using the CHARMM-GUI server. Each complex was placed inside a rectangular box with a buffer distance of 10 in each direction and solvated with
TIP3P water molecules. To neutralize the system’s charge for the Apigenin ligand, 5 Na + ions and 0 Cl– ions were added. Similarly, 5 Na + ions and 0 Cl– ions were added to neutralize the
system’s charge for the Apigenin-5-O-beta-d-glucopyranoside ligand, while 33 Na + ions and 0 Cl– ions were added for the L1 protein of human papillomavirus. Following this, 0.0 M of NaCl was
added to create an environment similar to cellular conditions for each complex. The complexes were subjected to structure minimization using the CHARMM36 force field. Each system was then
equilibrated at a temperature of 310 K for 5000 steps (10 PS) in the NPT ensemble during the production run, which lasted for 100 s. The position, velocity, and energy of the system were
recorded every 10 ps. Hydrogen atoms were constrained using the Lincs technique. A switching method of 12–14 was employed to calculate van der Waals forces, with a cutoff value of 1453,54.
Long-range electrostatic interactions were calculated using the particle mesh Ewald (PME) approach, with a maximum grid spacing of 1.2. The PME computations were conducted at each step,
without the use of a multiple-time stepping scheme. The temperature was maintained at 310 K, and the system size changes in the barostat were targeted at 1 bar. The integration time step was
set to 2 fs (0.002 = dt), and nsteps = 50,000,000 (50,000,000*2 = 100,000,000 ps = 100 ns). Post-simulation, the trajectories were analyzed using the VMD (University of Illinois at
Urbana-Champaign, Urbana, IL, USA) program, Bio3D, and QTGRACE, respectively, following re-centering of the simulation output55. After each MD simulation was completed, its trajectory was
analyzed to determine a variety of characteristics, such as the root mean square deviation (RMSD), root mean square fluctuation (RMSF), the radius of gyration (Rg), solvent-accessible
surface area (SASA), hydrogen bonds (H-bonds), principal component analysis (PCA), and dynamics cross-correlation map (DCCM). The resulting files were analyzed and visualized using xmgrace
(https://plasma-gate.weizmann.ac.il/Grace/), Bio3D, and VMD software56,57. BINDING FREE ENERGY CALCULATION Molecular mechanics/Poisson-Boltzmann surface area (M-PBSA) methodology provides a
comprehensive analysis of the quantitative assessment of the interaction mechanism between proteins and ligand molecules. The current investigation utilizes the MM-PBSA technique to evaluate
the binding affinity of a complex consisting of a protein and a ligand, to obtain a deeper understanding of the fundamental binding mechanism. The calculation of the van der Waals energy,
electrostatic energy, polar solvation energy, and binding energy was conducted for the Apigenin and Apigenin 5-O-Beta-D-Glucopyranoside complexes to forecast the overall ΔGbind. The
calculation of the binding free energy for the protein–ligand reaction was performed in the following manner: The equation ΔGbind = G complex–(G protein + G ligand) is utilized to determine
the total binding energy of the protein–ligand complex, where G protein denotes the binding energy of the protein and G ligand represents the binding energy of Apigenin and Apigenin
5-O-Beta-D-Glucopyranoside, as examined in this investigation. DENSITY FUNCTIONAL THEORY (QUANTUM MECHANICS) Density functional theory was used to conduct a quantum mechanical calculation on
the top twelve compounds (hits) from the virtual screening. The Gaussian 09W program was used for the calculations by optimizing the compounds' geometries at DFT/B3LYP/6-31G (d’p’)
levels58,59. The compounds were analyzed to determine their electron acceptor and electron donor properties by calculating the frontier orbital energies, including the highest occupied
molecular orbital (HOMO) and the lowest occupied molecular orbital (LUMO), as well as the energy gap and molecular electrostatic potential. These characteristics also provide information
regarding the chemical reactivity and stability of compounds60. RESULTS AND DISCUSSION LIPINSKI RULE AND PHARMACOKINETICS The complex balance of all molecular and structural characteristics
(molecular weight, lipophilicity, rotatable bonds, surface area, number of hydrogen bond acceptors and donors, bioavailability), as determined by the specific evolution of various
computational filters developed by Lipinski rule and it is included in the concept of drug-likeness. Our reported compound has a minimum violation of Lipinski rule except for two compounds
(CID 5319,484 and 129861,756) and these compounds have minimum bioavailability scores while others have very good bioavailability scores and most of them are about 0.55 or 55%
bioavailability (showing in Table 1). MOLECULAR DOCKING ANALYSIS AGAINST TARGETED RECEPTOR Initially, ADMET, PK, and drug-likeness were assessed, and it has been documented that most of the
molecules had accepted the guidelines of the Lipinski rule, PK, or the ADMET calculations. As a consequence of this, these compounds were molecularly docked and subjected to further
screening. The binding affinity is determined to measure how tightly inhibited or bonded the drugs with targeted protein are during the formation of complex structure61. It is said that
binding affinities of molecules greater than – 6.0 kcal/mol should be potential drug candidates62. After molecular docking, the result has been documented that the majority of the compounds
have been shown to have potent interactions and greater binding energies with both target proteins. In Table 2, the most effective compounds’ interactions with the HPV45 oncoprotein E7 (PDB
ID 2EWL) protein are reported Apigenin 4’-O-Rhamnoside, and Apigenin-4’-Alpha-L-Rhamnoside along with their docking scores − 6.9 kcal/mol, and − 6.7 kcal/mol against HPV45 oncoprotein E7
(PDB ID 2EWL). Besides, the binding affinities of the L1 protein of human papillomavirus (PDB ID 6L31) ranged from − 7.7 kcal/mol to − 9.3 kcal/mol, whereas molecules Apigenin and Apigenin
5-O-Beta-d-Glucopyranoside showed significant binding energy against L1 protein of human. Secondly, DNA polymerase plays an essential role in the therapeutic strategy of cancer and some
other disease by blocking or inhibiting DNA polymerases. Many hyperproliferative conditions, such as cancer, autoimmune diseases, and viral infections, are treated with drugs that block DNA
synthesis. So, the DNA polymerase theta is also included in this investigation, and perform molecular docking to determine the capability of whether the reported Apigenin derivatives can
inhibit the DNA polymerase theta or not and how much binding affinity is produced during binding with each other. This time, the binding affinity range is achieved − 7.5 kcal/mol to − 8.8
kcal/mol which represents that mentioned Apigenin derivatives should be inhibited the DNA polymerase theta (PDB ID 8E23). Besides, the vast majority of the Apigenin derivatives revealed more
potent interactions and also exhibited strong interactions and optimum binding affinity with the target. So, the Apigenin derivatives suggested performing wet-lab synthesis and then
evaluated on a biological or practical value, to establish as potential drug candidates. MOLECULAR DOCKING POSE AND ACTIVE SITE ANALYSIS Molecular docking pose and active site analysis has
been done by using discovery studio and Chimera X application. It helps to understand and visualize the specific amino acid residue where the ligand binds and formed a drug-protein in the
complex (Fig. 3). In this study, the best two complexes are visualized based on maximum binding energy. The first one is drug protein complex of HPV45 oncoprotein E7 (PDB ID 2EWL) with
Apigenin 4'-O-Rhamnoside where the active site are formed LEU A:45, LEU A:37, ASP A:33, ILE A: 23, LEU A: 25, THR A: 26, VAL A: 27 LEU A:45, LEU A:37, ASP A:33, ILE A: 23, LEU A: 25,
THR A: 26, VAL A: 27. Similarly, the second one is drug protein complex of L1 protein of human papillomavirus (PDB ID 6L31) with Apigenin. This time, the active residue or binding site are
located and form MET A:204, ILE A: 220, PHE A: 206, VAL A:216, PHE A: 201, ASN A:289, and VAL A: 268. THEORETICAL ADMET DATA ANALYSIS The predicted ADMET data of reported compounds are
available in Table 3. Few chemicals showed excellent human intestinal absorption and water solubility. This could help substances have an increased blood concentration for the best
biological activity. Additionally, these substances showed low blood–brain barrier (BBB) penetration, indicating a free from the creating CNS or neurotoxicity. Most of the reported ligands
were not causes of inhibitors of cytochrome P450 enzyme which indicate compounds are perfectly metabolized by cytochrome enzyme which is found in lever. Many mechanisms, primarily the liver
and kidney, are used to excretion of drug compounds from the body. Smaller drug molecules (< 300) were excreted from bile whereas bigger drug molecules (> 500) were removed from urine.
Between 300 and 500 molecular weights are excreted from bile and urine both63. For determining the excretion level of drug compounds, the total clearance rate of specific compounds is shown
in Table 3. Organic cation transporter 2 or OCT2 is another parameter that help to renal clearance of compounds. Where thus compounds did not expect to substrate on renal OCT2.
Unanticipated drug toxicity is a crucial factor in the failure of successful drug candidates and the withdrawal of marketed treatments. So, toxicity prediction of drug is one of the first
requirements for the development and discovery of the drugs. Therefore Table 4 present several toxicity parameters as: AMES toxicity, skin sensitization and hepatotoxicity. In this research
reported every article show positive result in ADMET prediction, which are non-toxic drug compound except one hepatotoxic compound. QSAR AND PIC50 CALCULATION Multiple linear regression
(MLR) analysis was used in quantitative structure–activity relationship (QSAR) investigations to determine the influence of compounds on pharmacological activity. The relationship between
biological activities and structural activities of chemical compounds has been calculated using the quantitative structure activities relationship (QSAR) method. It is discovered that
various compounds have distinct QSAR and pIC50 values, and the total value of the QSAR and pIC50 investigation fits all the requirements. The range of QSAR and pIC50 is determined to be
between 5.09 and 4.58, with 5.09 being the higher value and 4.58 being the lower value. According to Table 5, the predicted pIC50 indicates that these described compounds may have biological
significance against human papilloma virus. Following equation is applied which developed by another publication64. Here, \({\text{pIC5}}0 \, \left( {{\text{Activity}}} \right) \, = \, - {
2}.{768483965 } + \, 0.{133928895 } \times \, \left( {{\text{Chiv5}}} \right) \, + { 1}.{59986423 } \times \, \left( {{\text{bcutm1}}} \right) \, + \, \left( { - \, 0.0{23}0{9681}} \right)
\, \times \, \left( {{\text{MRVSA9}}} \right) \, + \, \left( { - \, 0.00{29461}0{1}} \right) \, \times \, \left( {{\text{MRVSA6}}} \right) \, + \, \left( {0.00{671218}} \right) \, \times \,
\left( {{\text{PEOEVSA5}}} \right) \, + \, \left( { - \, 0.{15963415}} \right) \, \times \, \left( {{\text{GATSv4}}} \right) \, + \, \left( {0.{2}0{7949857}} \right) \, \times \, \left(
{\text{J}} \right) \, + \, \left( {0.0{82568569}} \right) \, \times \, \left( {{\text{Diametert}}} \right).\) MOLECULAR DYNAMICS SIMULATION ANALYSIS The substantial root mean square
deviation (RMSD) value can be attributed to the protein's considerable size and its composition of five distinct chains. During the molecular dynamics’ simulation, an energy
minimization procedure was conducted at a time scale of 100 ns. Subsequently, the system was brought to a state of equilibrium. For energy minimization all three system, Using the steepest
descent algorithm, the energy of each system was minimized until the maximal force was less than 1,000,000 kj/mol/nm. This was performed to eliminate any steric conflicts within the system.
An isothermal-isochoric ensemble NVT (constant number of particles, volume, and temperature) and an isothermal-isobaric ensemble NPT (constant number of particles, pressure, and temperature)
were used to equilibrate each system. At 310 K and 1 bar pressure, the two types of ensemble equilibration methods stabilized the three systems. After the molecular dynamics simulation at
100 ns was completed, the PBC effect was eliminated by using the code gmx trjconv –f step5.xtc –o new.xtc –s step5.tpr –pbc mol –center –n index. ndx–ur compact in the Gromacs software, and
then it was checked in the VMD program and it was seen that there was no jump. ROOT-MEAN-SQUARE DEVIATION (RMSD) ANALYSIS To gain a better understanding of the dynamic behavior and
stability, the results of MD simulations for both the apo form and the ligand complex are investigated on a time scale of 100 ns. The MD simulation is carried out for a total of one hundred
nanoseconds, and the trajectories for the RMSD plot are displayed in Fig. 4. The colors in the figure are those associated with Apo protein at a time scale of 100 ns when it is complexed
with Apigenin and Apigenin-5-O-beta-d-glucopyranoside. The root means square deviation (RMSD) provides an interpretation regarding the extent to which a group of atoms deviates from the
appropriate original reference structure of a protein, ligand, or even a ligand–protein complex. A substantial amount of instability, which is related to changes within the conformation of
the molecule being researched, can be correlated with having high RMSD values. It was determined that the average RMSD for the Apigenin-6L31 and Apigenin-5-O-beta-d-glucopyranoside -protein
systems were 3.186 Å and 3.236 Å, respectively. The ligand-free protein, or apoprotein, has an RMSD value of 4.21 Å on average. For the apoprotein, we observed an abrupt increase in RMSD at
the outset, followed by a sudden decrease 2.5 ns later. Following the trajectories, Apo form increased progressively for 20 ns, after which the value exceeded the RMSD of Apigenin, which may
have occurred due to the higher occupancy of flexible loops in the C-terminal region and remained relatively stable until the end of the simulation. İn the case of the Apigenin complex
system, a comparable deviation pattern was observed. The RMSD of apoprotein was greater during the 20 ns and 40 ns, and for the 40–50 ns, the value was very close to that of the apoprotein
RMSD value. The RMSD of the Apigenin-6L31 system gradually increased and showed more fluctuations for 55 ns, after which the value attained its maximum at roughly 60 ns and declined to a
lower value at 78.7 ns of the trajectory. After this time, the observed slight increased and became stable at 100 ns with negligible fluctuations. Whereas, the RMSD for the backbone atom of
the Apigenin-5-O-beta-d-glucopyranoside complex concerning initial position quickly increased within the 20 ns run time and maintained a slightly non-significant fluctuation around 20 and 60
ns, where the following trajectories proceeded to drop slightly values till the end of the MD simulation. The average RMSD of the Apigenin-5-O-beta-D-glucopyranoside –6L31 is 0.82 greater
than that of apoproteins; however, after 65 ns, the average RMSD value is less than that of apoproteins. In agreement with the above observation, it can be concluded that Apigenin complexes
are very stable, as seen by the low RMSD value of Apigenin-5-O-beta-D-glucopyranoside − 6L31 after 60 ns and the constant RMSD value of Apigenin being very near to that of the apoprotein
throughout the 100 ns MD simulation. Consequently, the RMSD of the Apigenin-6L31 and Apigenin-5-O-beta-D-glucopyranoside − 6L31 systems may suggest that they did not endure substantial
conformational changes during the MD simulation. The RMSD histograms provided further evidence that the stability of the protein and ligand in the simulated system was seen and confirmed
(Fig. 5A–C). ROOT-MEAN-SQUARE FLUCTUATION (RMSF) The RMSF values are plotted to comprehend the residue-wise fluctuation between the apo and ligand complexes. The dynamic mobility in the loop
sections is demonstrated by the RMSD deviation for the apo and complexes. For understanding the residues that participated in the causative factors for fluctuations, the RMSF plot is
provided in Fig. 6. The RMSF value was utilized to identify the protein's hard and flexible regions. This validation criterion for structural variability in the ligand–protein complex
highlights the importance of specific protein residues in these structural shifts. Using a timescale of 100 ns, the amino acid at each position is calculated for its deviation value. For
apoprotein, the amino acid position from 407 to 416 exhibits a deviation of up to 15 Å, whereas other amino acids exhibit deviations of 1 to 5 Å. The deviation that occurs in the 407–416th
position and 1–5 A values may be the functional reason for the drift in apoprotein RMSD at 80th and 20 ns. In the process of comparing the values of the apo RMSF to those of the complex
protein RMSF, it has been observed that Apigenin-6L31 demonstrates greater deviations than the other ligand complex. The amino acid positions from 55 to 57th, 131st to 137th, 173rd to 178th,
347th to 354th, and 417th to 418th have deviations ranging from 5 to 10. Apigenin-6L31’s RMSD deviates between the 55th and 60th nanoseconds as a result of these positional amino acid
fluctuations. Similarly, another compound, Apigenin-5-O-beta-D glucopyranoside-6L31 also shows fluctuations in the same regions, only the amino acids between 277 and 280 exhibited higher
RMSF value, which confirms the greater RMSD value between 40 and 60 ns in the RMSD plot. Higher RMSF values are indicative of a more flexible protein structure. The protein–ligand system
produced RMSF values that were lower. RADIUS OF GYRATION (RG) ANALYSIS During the entirety of the simulation, the Rg parameter determines how compact a structure is. An increase in RoG
values suggests a reduction in the compactness of the protein structure, indicating increased flexibility and decreased stability. The Rg-time fluctuations were observed to be nearly
constant within the acceptable range, primarily maintained between 2.8 A and 2.6 A, indicating that the protein–ligand complexes undergo stable conformational changes. Compared to
Apigenin-6L31 and apo_6L31, the radius of gyration was smallest for the Apigenin-5-O-beta-D glucopyranoside-6L31 complex (Fig. 7). According to the findings that were obtained, the
Apigenin-5-O-beta-D glucopyranoside -6L31 complex was able to maintain a higher degree of stability during the simulation and bind successfully with the ligand. The trajectory of both
proteins was used to produce a plot known as a solvent-accessible surface area (SASA), which was then used to research the proportion of each system's surface area that can be reached
by the solvent (Fig. 7). SOLVENT ACCESSIBLE SURFACE AREA (SASA) ANALYSIS The information obtained from SASA will be useful for analyzing whether the ligand is kept inside the shallow binding
pocket or whether it is expelled from the binding cavity. From Fig. 8, the SASA for the Apo_6L31, Apigenin-6L31 complex, and Apigenin-5-O-beta-d glucopyranoside − 6L31 complex, where the
average SASA for the native protein was calculated to be 265.49, 261.037, and 255.149 nm2, was determined to be 265.49, 261.037, and 255.149 nm2, respectively. It has been shown that the
Apigenin-5-6L31 complex displays a lower SASA in comparison to the Apo_6L31 and Apigenin-6L31 complexes; this indicates that the Apigenin-5-O-beta-D glucopyranoside -6L31 complex is
responsible for inducing conformational alterations. HYDROGEN BOND ANALYSIS Throughout the simulation, and in addition to the RMSD and SASA analyses, we also examined the stability of the
hydrogen bonds (H-bonds) that are present in protein–ligand complexes. Understanding the connections between biomolecules necessitates a geometrical analysis of hydrogen bonding. Hydrogen
bonds are an important interaction in maintaining the structural integrity of biomolecules. Also, During MD modeling, the creation of H-bonds is an essential component in maintaining the
stability of the complexes, Throughout the entirety of the course of the MD simulation, it was discovered that the number of H-bonds that were present in the ligand-bound states was
constantly changing, as shown in Fig. 9. The total number of H-bonds that were found between Apigenin and the protein during the MD simulation was 19, whereas the number of H-bonds that were
found between Apigenin-5-O-beta-D glucopyranoside and the protein was a total of 8. It is evident from the graph that the Apigenin complex has more hydrogen bonds throughout the duration of
the simulation, whereas the Apigenin-5-O-beta-d glucopyranoside -6L31 complex has fewer hydrogen bonds. In the instance of the Apigenin-5-O-beta-d glucopyranoside-6L31 mutant, it was
discovered that there was a decrease in the number of hydrogen bonds when compared to Apigenin. Greater binding affinity is correlated with an increase in the number of hydrogen bonds formed
and their duration. In addition, utilizing H-bond occupancy allowed for the identification of vital residues that were involved in the creation of H-bonds for ligand recognition. Using the
VMD "Hydrogen bonds" tool, it was useful to explore the established ligand–protein hydrogen bond interactions and their relative frequencies65. The cut-off values for hydrogen bond
(Donor H. Acceptor) distance and angle were assigned at 3.0 Å and 20°, respectively66. Apigenin did not maintain all of the H-bonds that were detected in the docked complex, except Asn289;
however, it did form additional contacts with Ile220, Asp215, Thr199, Cys225, Tyr147, Thr223, Thr224, Arg259, Gly264, Val267, and Glu265. Apigenin-5 maintained only its interactions with
Asn289 and developed interactions with Tyr287, Phe201, Gly200, Trp165, and Tyr227 (Fig. 9), and Table 6 is displayed H-bond occupancy. CONTACT FREQUENCY (CF) ANALYSIS To, Fig. 10 depicts the
results of a contact frequency (CF) analysis performed with the contact Freq module on VMD and a cut-off of 4 to further evaluate the binding between 6L31 and the ligands. Phe206, Phe201,
Met204, Val216, Asp215, Arg259, Cys225, Val288, Ile220, Thr199, Met145, Gly202, Val267, Thr224, Leu271, Pro268 and Pro266, Hsd164, Tyr147, and Val263 had the highest CF during the
simulation. Apigenin exhibited a higher overall contact frequency with these residues than Apigenin-5. Moreover, the CFs of Phe201, Val288, and Gly202 with Apigenin and Apigenin-5 were quite
distant. The hydrogen bonds formed between protein and apigenin ligand were not the same as those formed between apigenin-5 and protein, except asn289. In addition, van der Waals
interactions were observed between Phe206, Phe201, Val216, Ile220, and Val288 with both ligands, all of which were identified in the presented high CF. Based on the H-bond analysis, we can
conclude that Apigenin complex binds to protein active sites more efficiently and tightly than Apigenin-5-6L31. Additionally, the presence of hydrogen bonds between 6L31 and Apigenin
derivatives has helped to strengthen the binding, which has contributed to the simulation's success in maintaining its stability (Fig. 10). PRINCIPAL COMPONENT ANALYSIS (PCA) Principal
Component Analysis (PCA) is a useful technique for extracting crucial information from Molecular Dynamics (MD) trajectories by modifying global slow motions from local fast motions. It was
utilized to model the significant dynamics of both the complex systems and the apo protein to investigate the nature of the interaction between the statistically significant conformations
that were found along the trajectory. The complex's essential variations were captured by arranging the principal components as eigenvectors according to their variance. Eigenvalue rank
plots display the fraction of total variation explained by each component. The PCA plots for the c-alpha backbone of the protein in the complex of Apigenin, Apigenin-5-O-beta-D
glucopyranoside, and 6L31-apo are shown in Fig. 11. Predicting the significant motions in the trajectory is useful. PCA was performed using RStudio and Bio3d67. Dynamic simulations are
essential to biological function, and PCA can isolate the most variable of these motions to investigate the conformational change of the systems, the PCA scatter plots of the 6L31 apo,
Apigenin, and Apigenin-5-O-beta-D glucopyranoside systems were generated by projecting the simulated trajectories of the protein systems into the two-dimensional subspace spanned by the
first three eigenvectors (PC1, PC2, and PC3). This allowed the conformational change of the systems to be investigated. Figure X displays the principal component analyses (PCA) that reveal
that the 6L31 apo, Apigenin, and Apigenin-5-O-beta-d glucopyranoside systems each contributed 44.6%, 31.29%, and 38.89 (14.29)% of the total variations, respectively. The Apigenin complex
was found to have the highest PC1 value (44.6%), which suggests that the complex has been subjected to a greater number of conformational changes. In contrast, the Apigenin-5 complex
exhibits less PC1 (31.29%), indicating that it has undergone a smaller conformational change. Moreover, the PC1 of the Apo structure was 38.9%, which is greater than the Apigenin-5-O-beta-D
glucopyranoside complex, indicating that the binding of Apigenin-5-O-beta-d glucopyranoside stabilizes the Apo's conformational changes. Figure 11(a, b and c) shows the conformational
state of the three systems in the subspace as depicted by the principal component analysis scatter diagram, with the red dot representing the stable conformation, the blue dot representing
the unstable conformation, and the white dots representing the intermediate state between the three conformations. DYNAMIC CROSS-CORRELATION MATRIX (DCCM) ANALYSIS To investigate the effect
of Apigenin derivatives on the conformational motions of the 6L31 protein, DCCM analyses were undertaken on all C atoms in the 6L31 apo, the Apigenin complex, and the Apigenin-5-O-beta-D
glucopyranoside complex system using 100 ns simulated trajectories (Fig. 12a, b and c). The DCCM exhibited a comprehensive correlation, encompassing a range of values from − 1.0 to 1.0, with
the former indicating a dark purple hue and the latter indicating a dark blue hue. It was determined that different shades of color correspond to varying degrees of correlation between
residues, with the deeper the color indicating a larger degree of association. The observed correlation coefficient, ranging from − 1 to 1, indicated that residues exhibited either a
positive or negative relationship in their movements. A positive correlation indicated that residues moved in the same direction, while a negative correlation indicated that residues moved
in opposite directions. Upon analyzing the DCCM diagrams of the three systems, it was observed that the correlated movements exhibited by each system were notably distinct. In contrast to
the Apigenin complex system, the collective movements that exhibit positive correlation in the entire Apigenin-5-O-beta-D glucopyranoside complex remained relatively stable, while the
movements that display negative correlation experienced a notable increase. The correlated movements of the Apigenin-5-O-beta-d glucopyranoside complex exhibit significant changes upon
ligand binding, particularly in marked areas denoted by black dashed boxes. THE BINDING FREE ENERGY ESTIMATION The MM/PBSA approach is a noteworthy technique utilized for the computation of
the binding free energy of protein–ligand complexes. The MM-PBSA method was employed to determine the binding free energy of the final 20 compounds based on the molecular dynamics (MD)
trajectories. The value of ΔG is determined by the collective impact of diverse protein–ligand interactions, including but not limited to van der Waals energy (ΔEvdW), electrostatic energy
(ΔEele), and EPB (electrostatic contribution to solvation-free energy by Poisson-Boltzmann) energy (Fig. 13). The binding energies of the Apigenin complex is − 29.61 kj/mol found in whereas
for Apigenin-5-O-beta-d glucopyranoside complex is − 0.13 kJ/mol. The Apigenin, exhibited ∆VDW (− 35.52 kcal/mol), ∆EEL (− 23.39 kcal/mol), and ∆EGB (32.49 kcal/mol), while compound
Apigenin-5 11 reflected ∆VDW (− 0.40 kcal/mol), ∆EEL (0.09 kcal/mol) and ∆EGB (0.29 kcal/mol) energies of completely different. The MM-PBSA analysis yielded findings indicating that Apigenin
exhibited robust binding energy and greater stability. The validation of the outcomes of the molecular docking and MD simulations were carried out through the binding free energy
calculation. DYNAMIC BEHAVIOR AND CONFIRMATIONAL CHANGE OF PROTEIN–LIGAND COMPLEX To understand the dynamic structural evolution of the L1 protein of human papillomavirus (PDB ID
6L31)-ligand throughout the 100 ns simulation time frame, nine snapshots at every 10 ns have been taken. Additionally, it has been shown that the ligands remain entirely attached to the
inhibitory site without undergoing any structural alteration, suggesting that they are quite stable (Fig. 14). FRONTIER MOLECULAR ORBITAL ANALYSIS (FMO) The present investigation employed
the Density Functional Theory (DFT) methodology based on quantum mechanics to compute the HOMO and LUMO energy of twelve compounds. The outcome of this analysis is depicted in Fig. 8. The
frontier orbitals, specifically the highest occupied molecular orbital (HOMO) and the lowest occupied molecular orbital (LUMO) can be utilized to characterize the reactivity of chemical
species68. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are utilized to characterize the electron-donating and accepting properties of
chemical compounds. An additional parameter that warrants consideration is the energy gap, denoting the disparity between the highest occupied molecular orbital (HOMO) and the lowest
unoccupied molecular orbital (LUMO) energies. This differential is indicative of intramolecular charge transfer and kinetic stability. Compounds possessing a significant energy gap exhibit
reduced chemical reactivity and heightened kinetic stability. In contrast, individuals possessing a narrow energy gap exhibit heightened reactivity and diminished kinetic stability. In this
study, the HOMO and LUMO energies of twelve compounds were calculated using the quantum mechanical Density Functional Theory (DFT) method, and the result is shown in Fig. 15. CHEMICAL
REACTIVITY AND MOLECULAR PROPERTIES ANALYSIS According to the findings, the compound Apigenin-7-O-Methyl Glucuronate exhibited the smallest energy gap (ΔE) in comparison to the other
compounds. This indicates a heightened level of chemical reactivity and significant intramolecular charge transfer from an electron donor (HOMO) to an electron acceptor (LUMO) group. The
present study examined two compounds, Apigenin and apigenin-5, and found that Apigenin exhibits a comparatively slightly lower energy gap than Apigenin 5-O-Beta-D-Glucopyranoside. A molecule
with a larger HOMO–LUMO energy gap indicates high chemical inertness and instability69. The principal factor behind this phenomenon is the obstruction of the electronic transition, which is
caused by a significant energy differential between the ground state and the excited state. Typically, a molecule exhibiting a small HOMO–LUMO gap indicates high stability. Based on the
findings presented in Table 7, it can be observed that the HOMO–LUMO gaps of all the chemicals under investigation fall within the range of 3.960 eV to 4.079 eV. Furthermore, the data
indicates that the order of the energy gap follows a descending pattern as follows: 10 > 9 > 6 > 2 > 7 > 1 > 8 > 5 > 3 > 4 > 12 > 11. The value of the
softness is displayed in Table 7. It is essential to keep in mind that the disintegration time required for an element will be shorter and that it will deteriorate at a faster pace than that
of other elements if its softness level is larger than a tiny value. On the other hand, the property of hardness is a fundamental characteristic of a substance, and its quantification
serves as an indicator of its durability. Typically, compounds with higher hardness values exhibit greater resistance to alterations in electron configuration at the molecular level. Our
reported molecules have shown 11 > 12 > 04 > 03 > 08 > 05 > 01 > 07 > 02 > 06 > 09 > 10, which means compounds 11, 12, and 04 will more rapidly disintegrate
compared to the other molecules. Again, the hardness is always opposite to softness, and the hardness values are reported as 10 > 09 > 06 > 02 > 07 > 01 > 05 > 08 >
03 > 04 > 12 > 11 in our studies, which indicates that the compounds 11, 12 and 04 are lower hardness and ultimately disintegrate quickly. CONCLUSION The effectiveness of Apigenin
derivatives has been utilized in this current investigation as the proposed compounds for the novel treatments for HPV-associated cervical cancer and the DNA polymerase theta since there is
no targeted therapy for them. This research gap encourages our research team to develop an urgent search for the potential molecules against them with novel modes of action. So, this in
silico study has been performed to screen potential drug candidates from a series of Apigenin derivatives with significant pharmacological properties. This current investigation also
includes the pharmacokinetic properties, drug-likeness, ADMET profiles, molecular docking, molecular dynamic simulation, PCA, DCCM DFT, and QSAR. The molecular dynamics simulation, and
molecular docking, methods were employed to prove the binding affinities against targeted receptors and the stability of the compounds. The results showed that all the derivatives of the
Apigenin molecule are drug-like, and promising hydrogen bonding was reported, exhibiting remarkably inhibitory capability for each of the targeted receptors and favorable binding energies.
The favorable rate-determining binding affinities across targeted protein ranges are − 7.1 kcal/mol to − 9.3 kcal/mol. It is also noted that Apigenin 5-O-Beta-d-Glucopyranoside was reported
maximum affinities (− 9.3 kcal/mol) against the L1 protein of human papillomavirus (PDB ID 6L31). Finally, our investigation found that the Apigenin derivatives should be suggested as a
novel compound against HPV-associated cervical cancer and the DNA polymerase theta. It is kept in mind that these advanced computational studies are provided the potential activity
theoretically. Further wet lab experiments should be conducted to validate this effect in vitro, in vivo, pre-clinical, and clinical trials. LIMITATIONS OF THE STUDY It is a theoretical
investigation; to validate this investigation, and develop newer and safer drugs from the synthetic sources, these derivatives must be carried out from computational to (in vitro and in
vivo), preclinical and clinical trials, to find out their practical value. DATA AVAILABILITY All data generated or analyzed during this study are included in this published article.
ABBREVIATIONS * ADMET: Absorption, distribution, metabolism, excretion and toxicity * ADP-ribose: Adenosine diphosphate ribose * ARG: Arginine * ASN: Asparagine * ASP: Aspartic acid *
ATPase: Adenosine diphosphatase * BBB: Blood brain barrier * Bp: Base pairs * BRCA1: Breast cancer 1 * BRCA2: Breast cancer 2 * CHARMM36: Chemistry at harvard macromolecular mechanics * CYS:
Cysteine * DCCM: Dynamic cross-correlation matrix Bio3D * DFT: Density functional theory * _DNA_ : Deoxyribonucleic acid * ELISA: Enzyme linked immunosorbent assay * EPB: Export promotion
bureau * FMO: Frontier molecular orbitals * GLN: Glutamine * GLU: Glutamic acid * GLY: Glycine * GNU: General public license * GROMACS-2019: GROningen machine for chemical simulations-2019 *
H-bond: Hydrogen bond * HIS: Histidine * HOMO: Highest occupied molecular orbital * HPV: Human papillomavirus * HR: Homologous recombination * HRR: Homologous recombinational repair * ILE:
Isoleucine * kcal/mol: Kilo calorie mole * LCR: Long control region * LEU: Leucine * LUMO: Lowest occupied molecular orbital * LYS: Lysine * MDs: Molecular dynamics simulation * MET:
Methionine * MLR: Multiple linear regression * MMEJ: Microhomology mediated end joining * MM-PBSA: Molecular mechanics Poisson-Boltzmann surface area * OCT2: Organic cation transporter 2 *
ORFs: Open reading frames * P97: Protein 97 * PARPi: Poly adenosine diphosphate ribose polymerase * PCA: Principal component analysis * PCR: Polymerase chain reaction * PHE: Phenylalanine *
pkCSM: Predicting small molecule pharmacokinetic and toxicity properties using graph-based signatures * PME: Particle mesh Ewald * Pol θ: DNA polymerase theta * PRO: Proline * QSAR:
Quantitative structure activity relationship * RCSB: Research collaboratory for structural bioinformatics * RMSD: Root Mean square deviation * RMSF: Root Mean square fluctuation * RoG
profiles: Radius of gyration * RPA: Replication protein A * SASA: Solvent accessible surface area * SER: Serine * SMILES: Simplified molecular input line entry system * THR: Threonine *
TIP3P: Transferable intermolecular potential with 3 point * TRP: Tryptophan * VAL: Valine * VMD: Visual molecular dynamics REFERENCES * Poniewierza, P. & Panek, G. Cervical cancer
prophylaxis—State-of-the-art and perspectives. _Healthcare_ 10, 1325 (2022). PubMed Central PubMed Google Scholar * Rahib, L., Wehner, M. R., Matrisian, L. M. & Nead, K. T. Estimated
projection of US cancer incidence and death to 2040. _JAMA Netw. Open_ 4, e214708–e214708 (2021). PubMed Central PubMed Google Scholar * Chan, C. K., Aimagambetova, G., Ukybassova, T.,
Kongrtay, K. & Azizan, A. Human papillomavirus infection and cervical cancer: Epidemiology, screening, and vaccination—Review of current perspectives. _J. Oncol._ 2019, 1–11 (2019).
Google Scholar * Jalil, A. A. T. Epidemiology of Cervical cancer and high risk of human papilloma virus in patient. _ББК 28.6 З,_ 85, 7. * Kanda, T. & Kukimoto, I. Human papillomavirus
and cervical cancer. _Uirusu_ 56, 219–230 (2006). CAS PubMed Google Scholar * Gravitt, P. E. _et al._ A comparison between real-time polymerase chain reaction and hybrid capture 2 for
human papillomavirus DNA quantitation. _Cancer Epidemiol. Biomark. Prev._ 12, 477–484 (2003). CAS Google Scholar * Kelesidis, T. _et al._ Human papillomavirus (HPV) detection using in situ
hybridization in histologic samples: Correlations with cytologic changes and polymerase chain reaction HPV detection. _Am. J. Clin. Pathol._ 136, 119–127 (2011). PubMed Google Scholar *
Watts, D. H. _et al._ Low risk of perinatal transmission of human papillomavirus: Results from a prospective cohort study. _Am. J. Obstet. Gynecol._ 178, 365–373 (1998). CAS PubMed Google
Scholar * Einstein, M. H. & Goldberg, G. L. Human papillomavirus and cervical neoplasia. _Cancer Investig._ 20, 1080–1085 (2002). Google Scholar * Thompson, L. H. & Schild, D.
Homologous recombinational repair of DNA ensures mammalian chromosome stability. _Mutat. Res. Fundam. Mol. Mech. Mutagenes._ 477, 131–153 (2001). CAS Google Scholar * Hoppe, M. M., Sundar,
R., Tan, D. S. P. & Jeyasekharan, A. D. Biomarkers for homologous recombination deficiency in cancer. _J. Natl. Cancer Inst._ 110, 704–713 (2018). PubMed Google Scholar * Lord, C. J.
& Ashworth, A. J. S. PARP inhibitors: Synthetic lethality in the clinic. _Science_ 355, 1152–1158 (2017). ADS PubMed Central CAS PubMed Google Scholar * Bryant, H. E. _et al._
Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. _Nature_ 434, 913–917 (2005). ADS CAS PubMed Google Scholar * Farmer, H. _et al._ Targeting
the DNA repair defect in BRCA mutant cells as a therapeutic strategy. _Nature_ 434, 917–921 (2005). ADS CAS PubMed Google Scholar * Ledermann, J. _et al._ Olaparib maintenance therapy in
platinum-sensitive relapsed ovarian cancer. _N. Engl. J. Med._ 366, 1382–1392 (2012). CAS PubMed Google Scholar * Coleman, R. L. _et al._ Rucaparib maintenance treatment for recurrent
ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. _Lancet_ 390, 1949–1961 (2017). PubMed Central CAS PubMed
Google Scholar * Mirza, M. R. _et al._ Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. _N. Engl. J. Med._ 375, 2154–2164 (2016). CAS PubMed Google Scholar
* Litton, J. K. _et al._ Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. _N. Engl. J. Med._ 379, 753–763 (2018). PubMed Central CAS PubMed Google Scholar
* Zhou, J. _et al._ A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. _Nat. Cancer_ 2, 598–610 (2021). PubMed Central CAS PubMed
Google Scholar * Carvajal-Garcia, J., Crown, K. N., Ramsden, D. A. & Sekelsky, J. DNA polymerase theta suppresses mitotic crossing over. _PLoS Genet._ 17, e1009267 (2021). PubMed
Central CAS PubMed Google Scholar * Wood, R. D. & Doublié, S. DNA polymerase θ (POLQ), double-strand break repair, and cancer. _DNA Repair_ 44, 22–32 (2016). PubMed Central CAS
PubMed Google Scholar * Seki, M., Marini, F. & Wood, R. D. POLQ (Pol θ), a DNA polymerase and DNA-dependent ATPase in human cells. _Nucleic Acids Res._ 31, 6117–6126 (2003). PubMed
Central CAS PubMed Google Scholar * Ceccaldi, R. _et al._ Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. _Nature_ 518, 258–262 (2015). ADS PubMed
Central CAS PubMed Google Scholar * Mateos-Gomez, P. A. _et al._ Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. _Nature_ 518, 254–257 (2015). ADS PubMed
Central CAS PubMed Google Scholar * Chan, S. H., Yu, A. M. & McVey, M. Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila.
_PLoS Genet._ 6, e1001005 (2010). PubMed Central PubMed Google Scholar * Higgins, G. S. & Boulton, S. J. Beyond PARP—POLθ as an anticancer target. _Science_ 359, 1217–1218 (2018). ADS
CAS PubMed Google Scholar * Rivera, A. & Tyring, S. K. Therapy of cutaneous human papillomavirus infections. _Dermatol. Ther._ 17, 441–448 (2004). PubMed Google Scholar * Kumer,
A. _et al._ Investigation of the new inhibitors by sulfadiazine and modified derivatives of α-D-glucopyranoside for white spot syndrome virus disease of shrimp by in silico: Quantum
calculations, molecular docking, ADMET and molecular dynamics study. _Molecules_ 27, 3694 (2022). PubMed Central CAS PubMed Google Scholar * Rahman, M. M. _et al._ Use of computer in
drug design and drug discovery: A review. _Int. J. Pharm. Life Sci._ https://doi.org/10.3329/ijpls.v1i2.12955 (2012). Article Google Scholar * Lowy, D. R. & Schiller, J. T.
Prophylactic human papillomavirus vaccines. _J. Clin. Investig._ 116, 1167–1173 (2006). PubMed Central CAS PubMed Google Scholar * Ferraro, C. T. L., Canedo, N. H. S., de Oliveira, S.
P., da Glória da Costa Carvalho, M. & Dias, E. P. HPV oral infection and proliferative epithelial associated lesions. _J. Bras. Patol. Med. Lab._ 47, 451–459 (2011). Google Scholar *
Cai, Q., Lv, L., Shao, Q., Li, X. & Dian, A. Human papillomavirus early proteins and apoptosis. _Arch. Gynecol. Obstet._ 287, 541–548 (2013). CAS PubMed Google Scholar * Doorbar, J.
Host control of human papillomavirus infection and disease. _Best Pract. Res. Clin. Obstet. Gynaecol._ 47, 27–41 (2018). PubMed Google Scholar * Pal, A. & Kundu, R. Human
papillomavirus E6 and E7: The cervical cancer hallmarks and targets for therapy. _Front. Microbiol._ 10, 3116 (2020). PubMed Central PubMed Google Scholar * Kruchinin, A. A. &
Makarova, A. V. Multifaceted nature of DNA polymerase θ. _Int. J. Mol. Sci._ 24, 3619 (2023). PubMed Central CAS PubMed Google Scholar * Bubenik, M. _et al._ Identification of RP-6685,
an orally bioavailable compound that inhibits the DNA polymerase activity of Polθ. _J. Med. Chem._ 65, 13198–13215 (2022). PubMed Central CAS PubMed Google Scholar * Cadoná, F. C. _et
al._ Natural product–based nanomedicine: Polymeric nanoparticles as delivery cargoes of food bioactives and nutraceuticals for anticancer purposes. In _Advances and Avenues in the
Development of Novel Carriers for Bioactives and Biological Agents_ (eds Cadoná, F. C. _et al._) 37–67 (Elsevier, 2020). Google Scholar * Sen, P. _et al._ Apigenin naturally occurring
flavonoids: Occurrence and bioactivity. _Pharm. Biosci. J._ https://doi.org/10.20510/ukjpb/4/i6/134666 (2016). Article Google Scholar * Wang, M., Firrman, J., Liu, L. & Yam, K. A
review on flavonoid apigenin: Dietary intake, ADME, antimicrobial effects, and interactions with human gut microbiota. _BioMed. Res. Int._ 2019, 1–18 (2019). CAS Google Scholar * Xu, L.
_et al._ The anticancer potential of apigenin via immunoregulation. _Curr. Pharm. Des._ 27, 479–489 (2021). CAS PubMed Google Scholar * Kim, S. _et al._ PubChem 2019 update: Improved
access to chemical data. _Nucleic Acids Res._ 47, D1102–D1109 (2019). PubMed Google Scholar * Ferreira, L. G., Dos Santos, R. N., Oliva, G. & Andricopulo, A. D. Molecular docking and
structure-based drug design strategies. _Molecules_ 20, 13384–13421 (2015). PubMed Central CAS PubMed Google Scholar * Ravi, L. & Kannabiran, K. A handbook on protein-ligand docking
tool: AutoDock 4. _Innov. J. Med. Sci._ 4, 28–33 (2016). Google Scholar * Ohlenschläger, O. _et al._ Solution structure of the partially folded high-risk human papilloma virus 45
oncoprotein E7. _Oncogene_ 25, 5953–5959 (2006). PubMed Google Scholar * Liu, X. _et al._ Neutralization sites of human papillomavirus-6 relate to virus attachment and entry phase in viral
infection. _Emerg. Microbes Infect._ 8, 1721–1733 (2019). PubMed Central PubMed Google Scholar * Eddershaw, P. J., Beresford, A. P. & Bayliss, M. K. ADME/PK as part of a rational
approach to drug discovery. _Drug Discov. Today_ 5, 409–414 (2000). CAS PubMed Google Scholar * Azzam, K. A. SwissADME and pkCSM webservers predictors: An integrated online platform for
accurate and comprehensive predictions for in silico ADME/T properties of artemisinin and its derivatives. _Kompleksnoe Ispolzovanie Mineralnogo Syra_ 325, 14–21 (2023). Google Scholar *
Gaviraghi, G., Barnaby, R. J. & Pellegatti, M. Pharmacokinetic challenges in lead optimization. In _Pharmacokinetic Optimization in Drug Research: Biological, Physicochemical, and
Computational Strategies_ (eds Testa, B. _et al._) 1–14 (Wiley, 2001). Google Scholar * Mortier, J. _et al._ The impact of molecular dynamics on drug design: Applications for the
characterization of ligand–macromolecule complexes. _Drug Discov. Today_ 20, 686–702 (2015). CAS PubMed Google Scholar * Lazim, R., Suh, D. & Choi, S. Advances in molecular dynamics
simulations and enhanced sampling methods for the study of protein systems. _Int. J. Mol. Sci._ 21, 6339 (2020). PubMed Central CAS PubMed Google Scholar * Yalcin-Ozkat, G. Molecular
modeling strategies of cancer multidrug resistance. _Drug Resist. Updates_ 59, 100789 (2021). CAS Google Scholar * Murail, S. Simulation of ligand binding to membrane proteins. In
_Membrane Protein Structure and Function Characterization: Methods and Protocols_ (ed. Lacapere, J.-J.) 359–381 (Springer New York, 2017). Google Scholar * Joung, I. S. & Cheatham, T.
E. III. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. _J. Phys. Chem. B_ 112, 9020–9041 (2008). PubMed Central CAS
PubMed Google Scholar * Wells, D. B. & Aksimentiev, A. Mechanical properties of a complete microtubule revealed through molecular dynamics simulation. _Biophys. J._ 99, 629–637 (2010).
ADS PubMed Central CAS PubMed Google Scholar * Nnyigide, O. S., Lee, S.-G. & Hyun, K. Exploring the differences and similarities between urea and thermally driven denaturation of
bovine serum albumin: Intermolecular forces and solvation preferences. _J. Mol. Model._ 24, 1–15 (2018). CAS Google Scholar * Rana, N. _et al._ Drug resistance mechanism of
m46i-mutation-induced saquinavir resistance in HIV-1 protease using molecular dynamics simulation and binding energy calculation. _Viruses_ 14, 697 (2022). PubMed Central CAS PubMed
Google Scholar * Banerjee, S., Majumder, K., Gutierrez, G. J., Gupta, D. & Mittal, B. Immuno-informatics approach for multi-epitope vaccine designing against SARS-CoV-2. _BioRxiv_
https://doi.org/10.1101/2020.07.23.218529 (2020). Article PubMed Central PubMed Google Scholar * Cao, W. _et al._ Theoretical study of a series of 4, 4′-azo-1 H-1, 2, 4-triazol-5-one
based nitrogen-rich salts as potential energetic compounds. _RSC Adv._ 8, 23805–23816 (2018). ADS PubMed Central CAS PubMed Google Scholar * Rajalakshmi, K., Gunasekaran, S. &
Kumaresan, S. Density functional theory, comparative vibrational spectroscopic studies, highest occupied molecular orbital and lowest unoccupied molecular orbital analysis of Linezolid.
_Indian J. Phys._ 89, 525–538 (2015). ADS CAS Google Scholar * Kumer, A., Sarker, M. N. & Sunanda, P. The theoretical investigation of HOMO, LUMO, thermophysical properties and QSAR
study of some aromatic carboxylic acids using HyperChem programming. _Int. J. Chem. Technol._ 3, 26–37 (2019). Google Scholar * Mehmood, A., Kaushik, A. C., Wang, Q., Li, C.-D. & Wei,
D.-Q. Bringing structural implications and deep learning-based drug identification for KRAS mutants. _J. Chem. Inf. Model._ 61, 571–586 (2021). CAS PubMed Google Scholar * Nath, A.,
Kumer, A., Zaben, F. & Khan, M. Investigating the binding affinity, molecular dynamics, and ADMET properties of 2, 3-dihydrobenzofuran derivatives as an inhibitor of fungi, bacteria, and
virus protein. _Beni-Suef Univ. J. Basic Appl. Sci._ 10, 1–13 (2021). Google Scholar * Shamsuddin, T., Hosen, M. A., Alam, M. S., Emran, T. B. & Kawsar, S. M. A. Uridine derivatives:
Antifungal, PASS outcomes, ADME/T, drug-likeliness, molecular docking and binding energy calculations. _Medicine_ 10, 1373–1386 (2021). Google Scholar * Siddikey, F., Roni, M., Kumer, A.,
Chakma, U. & Matin, M. Computational investigation of Betalain derivatives as natural inhibitor against food borne bacteria. _Curr. Chem. Lett._ 11, 309–320 (2022). Google Scholar *
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. _J. Mol. Gr._ 14, 33–38 (1996). CAS Google Scholar * Joshi, A. A., Narkhede, S. S. & Viswanathan, C. Design,
synthesis and evaluation of 5-substituted amino-2, 4-diamino-8-chloropyrimido-[4, 5-b] quinolines as novel antimalarials. _Bioorg. Med. Chem. Lett._ 15, 73–76 (2005). CAS PubMed Google
Scholar * Qureshi, R., Ghosh, A. & Yan, H. Correlated motions and dynamics in different domains of epidermal growth factor receptor with L858R and T790M mutations. _IEEE/ACM Trans.
Comput. Biol. Bioinf._ 19, 383–394 (2020). Google Scholar * Saha, S. K., Hens, A., Murmu, N. C. & Banerjee, P. A comparative density functional theory and molecular dynamics simulation
studies of the corrosion inhibitory action of two novel N-heterocyclic organic compounds along with a few others over steel surface. _J. Mol. Liq._ 215, 486–495 (2016). CAS Google Scholar
* Kumer, A., Sarker, M. N. & Paul, S. The thermo physical, HOMO, LUMO, vibrational spectroscopy and QSAR study of morphonium formate and acetate ionic liquid salts using computational
method. _Turk. Comput. Theor. Chem._ 3, 59–68 (2019). CAS Google Scholar Download references ACKNOWLEDGEMENTS The authors extend their appreciation to the Deputyship for Research &
innovation, “Ministry of Education” in Saudi Arabia for funding this research (IFKSUOR3-155-3). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Pharmacy, Faculty of Allied Health
Sciences, Daffodil International University, Birulia, Ashulia, Dhaka, 1216, Bangladesh Shopnil Akash & Md. Rezaul Islam * Department of Bioinformatics and Computational Biology,
Gaziantep University, Gaziantep, Turkey Imren Bayıl * Department of Biomedical Engineering, Faculty of Engineering & Technology, Islamic University, Kushtia, Bangladesh Md. Saddam
Hossain * Professor Joarder DNA and Chromosome Research Laboratory, Department of Genetic Engineering and Biotechnology, University of Rajshahi, Rajshahi, 6205, Bangladesh Md. Eram Hosen *
Department of Biology, Bahir Dar University, P.O.Box 79, Bahir Dar, Ethiopia Amare Bitew Mekonnen * Department of Food Science, Faculty of Agricultural and Food Sciences, Laval University,
2325, Quebec City, QC, G1V 0A6, Canada Hiba-Allah Nafidi * Department of Pharmaceutics, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia Yousef A. Bin Jardan * Department of
Chemistry and Biochemistry, Faculty of Medicine and Pharmacy, Ibn Zohr University, 70000, Laayoune, Morocco Mohammed Bourhia * Department of Pathology and Laboratory Medicine, Warren Alpert
Medical School & Legorreta Cancer Center, Brown University, Providence, RI 02912, United States Talha Bin Emran Authors * Shopnil Akash View author publications You can also search for
this author inPubMed Google Scholar * Imren Bayıl View author publications You can also search for this author inPubMed Google Scholar * Md. Saddam Hossain View author publications You can
also search for this author inPubMed Google Scholar * Md. Rezaul Islam View author publications You can also search for this author inPubMed Google Scholar * Md. Eram Hosen View author
publications You can also search for this author inPubMed Google Scholar * Amare Bitew Mekonnen View author publications You can also search for this author inPubMed Google Scholar *
Hiba-Allah Nafidi View author publications You can also search for this author inPubMed Google Scholar * Yousef A. Bin Jardan View author publications You can also search for this author
inPubMed Google Scholar * Mohammed Bourhia View author publications You can also search for this author inPubMed Google Scholar * Talha Bin Emran View author publications You can also search
for this author inPubMed Google Scholar CONTRIBUTIONS Conceptualization, writing the original draft, formal analysis: S.A., I.B., Md.S.H. Investigations, funding acquisition, resources,
project administration: S.A Md.R.I. and Md.E.H., A.B.M. Reviewing and editing, data validation, and data curation: H.-A.N., Y.A.B.J., M.B. Supervision: T.B.E and M.B. CORRESPONDING AUTHORS
Correspondence to Shopnil Akash, Amare Bitew Mekonnen or Talha Bin Emran. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION
PUBLISHER'S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article
is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in
this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's
Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Akash, S., Bayıl, I., Hossain, M.S. _et al._
Novel computational and drug design strategies for inhibition of human papillomavirus-associated cervical cancer and DNA polymerase theta receptor by Apigenin derivatives. _Sci Rep_ 13,
16565 (2023). https://doi.org/10.1038/s41598-023-43175-x Download citation * Received: 05 May 2023 * Accepted: 20 September 2023 * Published: 02 October 2023 * DOI:
https://doi.org/10.1038/s41598-023-43175-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Exploiting pathogenic Escherichia coli to model transmembrane receptor signallingMany pathogens deploy a sophisticated virulence effector repertoire to promote their colonization, entry, survival and d...
Unsafe Lead Levels Found At Three More CMS Schools | WFAE 90.7 - Charlotte's NPR News SourceEducation Unsafe Lead Levels Found At Three More CMS Schools WFAE | By WFAE Published December 11, 2018 at 4:56 PM EST F...
Sidewire is your hotline to political insight | techcrunchAiming to help people cut through all of the political noise, Sidewire, a political news analysis platform, is launching...
[image] [https://www.portaltemponovo.com.br/wp-content/uploads/2018/10/WhatsApp-Image-2018-10-19-at-2.13.56-PM-1.jpeg][image] [https://www.portaltemponovo.com.br/wp-content/uploads/2018/10/WhatsApp-Image-2018-10-19-at-2.13.56-PM-1.jpeg]...
Polysaccharides from pseudostellaria heterophylla modulate gut microbiota and alleviate syndrome of spleen deficiency in ratsABSTRACT _Pseudostellaria heterophylla_, also called Tai-zi-shen (TZS) in Traditional Chinese Medicine (TCM), is always ...
Latests News
Novel computational and drug design strategies for inhibition of human papillomavirus-associated cervical cancer and dna polymerase theta receptor byABSTRACT The present study deals with the advanced in-silico analyses of several Apigenin derivatives to explore human p...
Double and triple ionisation of isocyanic acidABSTRACT Double and triple ionisation spectra of the reactive molecule isocyanic acid (HNCO) have been measured using mu...
Fate of 1,078 candidates to be sealed tomorrow in PunjabAbout 200 companies of the central Para military forces, besides Punjab Police personnel, have been deployed to ensure f...
Not all cruise ships had coronavirus outbreaks. It came down to a 'stroke of luck'BARCELONA, Spain — For Spanish traveler Carlos Payá, being on an around-the-globe luxury cruise while the rest of the wo...
Emmerdale spoilers: robert sugden to kill victoria's rapist lee?Robert (played by Ryan Hawley) has been struggling to process his little sister Victoria’s (Isabel Hodgins) trauma on Em...