Water-accelerated π-stacking reaction in benzene cluster cation
Water-accelerated π-stacking reaction in benzene cluster cation"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Single molecule electron devices (SMEDs) have been widely studied through both experiments and theoretical calculations because they exhibit certain specific properties that general
macromolecules do not possess. In actual SMED systems, a residual water molecule strongly affects the electronic properties of the SMED, even if only one water molecule is present. However,
information about the effect of H2O molecules on the electronic properties of SMEDs is quite limited. In the present study, the effect of H2O on the ON-OFF switching property of
benzene-based molecular devices was investigated by means of a direct ab initio molecular dynamics (AIMD) method. T- and H-shaped benzene dimers and trimers were examined as molecular
devices. The present calculations showed that a H2O molecule accelerates the π-stacking formation in benzene molecular electronic systems. The times of stacking formation in a benzene dimer
cation (_n_ = 2) were calculated to be 460 fs (_H_2_O_) and 947 fs (_no-H_2_O_), while those in a trimer cation (_n_ = 3) were 551 fs (_H_2_O_) and 1019 fs (_no-H_2_O_) as an average of the
reaction time. This tendency was not dependent on the levels of theory used. Thus, H2O produced positive effects in benzene-based molecular electronics. The mechanism of π-stacking was
discussed based on the theoretical results. SIMILAR CONTENT BEING VIEWED BY OTHERS Σ–Σ STACKED SUPRAMOLECULAR JUNCTIONS Article 28 July 2022 APPROACHING ISOTROPIC CHARGE TRANSPORT OF N-TYPE
ORGANIC SEMICONDUCTORS WITH BULKY SUBSTITUENTS Article Open access 11 November 2021 ELECTRICAL CONTROL OF TRANSIENT FORMATION OF ELECTRON-HOLE COEXISTING SYSTEM AT SILICON
METAL-OXIDE-SEMICONDUCTOR INTERFACES Article Open access 31 October 2023 INTRODUCTION Molecular electronics is a new field of technology that is based on the applications of electronic
devices composed of organic molecules1,2,3,4,5,6,7. Although a variety of functional molecular devices such as molecular switches8,9,10 and molecular rectifiers11,12 have been prepared
successfully, the field of molecular electronics is still very fertile from a fundamental research perspective, allowing rapid progress to be made in device performance and reliability.
Among molecular electronic devices, single molecule (including small cluster)-electron devices (SMEDs) have been widely investigated, by experiments and theoretical calculations, in view of
certain specific properties that general macromolecules do not possess. For instance, slight changes in the molecular structure and constituent atoms of SMEDs can lead to large differences
in electron conductivity. Using density functional theory (DFT) calculations, Yang _et al_. investigated the effect of atom substitution on the electron transport properties in
dehydroazulene13. They suggested that different substitution positions of fluorine atoms in the molecule had a significant influence on the switching property. In actual SMED systems, a
residual water molecule may strongly affect the electronic properties, even if only one water molecule is present. However, information about the effect of H2O molecules on the electronic
properties of SMEDs is quite limited. Recently, Li _et al_. investigated the effect of H2O on the conductance of a single molecular junction composed of thiolated arylethynylene with a
9,10-dihydroanthracene core (denoted as TADHA)14. These authors showed that H2O suppressed the conductance of the molecular junction if they adsorbed on the terminal sulfur atoms. The
circuit of electron transport in Na-NTCDA (1,4,5,8-naphthalene-tetracarboxylic-dianhydride) was easily destroyed by a single water molecule15. In molecular devices comprising biphenyl
molecules16, H2O strongly affected the hole transport properties. Thus, previous reports have shown that the H2O molecule generally produces negative effects on molecular
electronics17,18,19,20. In the present study, the effect of a single water molecule on the ON-OFF switching property of benzene-based molecular devices was investigated using the direct ab
initio molecular dynamics (AIMD) method21,22,23. The small-sized benzene clusters have the ability to act as an ON-OFF switching element24,25,26. It is known that a T-shaped benzene dimer is
drastically changed to the π-stacking form after hole capture, while an H-shaped benzene trimer is changed to the double π-stacking form. In this work, we mainly focused on the effect of
H2O on the time-scale of π-stacking formation in benzene clusters. COMPUTATIONAL DETAILS STATIC DENSITY FUNCTIONAL THEORY (DFT) CALCULATIONS The geometries of a T-shaped benzene dimer,
H-shaped benzene trimer, hydrated benzene dimer, and hydrated benzene trimer were fully optimized using the CAM-B3LYP/6-311++G(d,p) method. The atomic and molecular charges were calculated
using natural population analysis (NPA). The standard Gaussian 09 program package was used for all static ab initio calculations27. DIRECT AIMD CALCULATIONS The trajectory of (Bz)n+
following the ionization of (Bz)n (_n_ = 2) was calculated using direct AIMD21,22,23 at the CAM-B3LYP/6-31 G(d) level under the assumption of vertical ionization in the neutral state. The
optimized structure obtained at the CAM-B3LYP/6-311++G(d,p) level was chosen as the initial geometry of (Bz)n+ at time zero. The trajectory calculation of (Bz)n+ was performed using the
condition of constant total energy. The velocity Verlet algorithm was used with a time step of 0.5 fs to solve the equation of motion for the system. The drifts in the total energies in all
trajectory calculations were less than 0.01 kcal/mol. Similar calculations were carried out for hydrated benzene dimer and trimer systems. To investigate the effect of the initial structures
on the reaction mechanism and time-scale of stacking formation in (Bz)2+ and (Bz)2+-H2O, the initial geometries were generated by direct AIMD calculations under a constant temperature
condition28,29. The temperature was chosen as 10 K. First, direct AIMD calculations of the neutral systems, (Bz)2 and (Bz)2-H2O, were carried out at the CAM-B3LYP/6-311++G(d,p) level at 10
K. Second, the geometries and velocities of the atoms were selected from the simulations. Next, direct AIMD calculations were carried out for cation systems at the CAM-B3LYP/6-31G(d) level.
Twelve trajectories were run from the selected points. In addition to the 10 K simulation, the optimized structures of the cations calculated by several levels of theory were examined as the
initial structures in the direct AIMD calculations. The basis sets used were (A) 6-311++G(2d,p), (B) 6-311++G(2d,2p), (C) 6-311++G(2df,2p), (D) 6-311++G(2df,2pd), (E) 6-311++G(3df,2pd), and
(F) 6-311++G(3df,3pd). The trajectories began from these optimized structures, and the times of π-stacking formation were calculated. The dependence of the reaction time on the functional
used for DFT calculations was assessed by using APFD, B3LYP, M052X, and M062X functionals. Note that the dependence on the type of functional was significantly small, as shown in later
sections. RESULTS ELECTRONIC STATES OF (BZ)N AND (BZ)N-H2O (_N_ = 2 AND 3) The structures of the benzene (Bz)n and hydrated benzene (Bz)n-H2O dimers and trimers (_n_ = 2 and 3) were fully
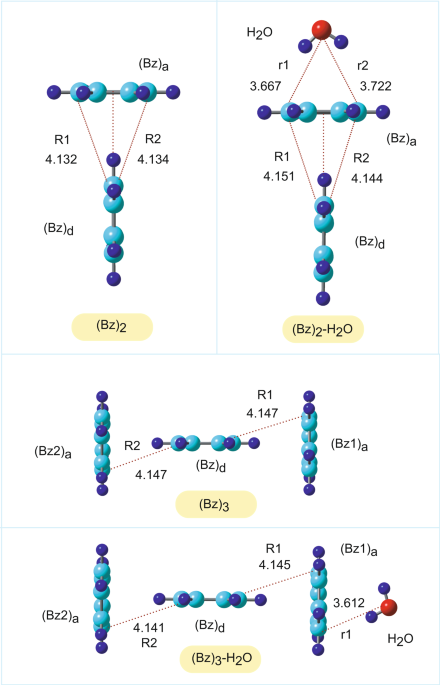
optimized at the CAM-B3LYP/6-311++G(d,p) level. The optimized structures are given in Fig. 1. The benzene dimer (_n_ = 2) is composed of proton donor and acceptor benzene molecules, which
are denoted as (Bz)d and (Bz)a, respectively. The intermolecular carbon-carbon distances (Å) were (R1, R2) = (4.132, 4.134) in (Bz)2 and (4.151, 4.144) in (Bz)2-H2O, indicating that the
effect of H2O on the structure was negligibly small, although the distance increased slightly as a result of the interaction with H2O. In (Bz)2-H2O, the dipole of H2O was oriented toward the
center of mass in (Bz)a, while the distances of H2O from (Bz)a (in Å) were (r1, r2) = (3.667, 3.722). The NPA charges are given in Table 1. The NPA molecular charges on (Bz)d and (Bz)a in
(Bz)2 were −0.002 and +0.002, respectively, indicating that the electron transfer from (Bz)a to (Bz)d was negligibly small. In the case of (Bz)2-H2O, the NPA molecular charges on (Bz)d,
(Bz)a, and H2O were −0.002, 0.000, and +0.002, respectively. The charge distribution on the neutral benzene dimer was nearly unaffected by the interaction with H2O. The benzene trimer (_n_ =
3) is composed of one proton donor and two acceptor benzene molecules, which are denoted as (Bz)d, (Bz1)a, and (Bz2)a, respectively. The intermolecular distances (R1, R2) were (4.147,
4.147) in (Bz)3 and (4.145, 4.141) in (Bz)3-H2O. The dipole of H2O orients benzene (Bz1)a. The NPA charges of (Bz)3 and (Bz)3-H2O are given in Table 2. The effects of H2O on the electronic
states were negligibly small in the neutral state for _n_ = 2 and 3. ELECTRONIC STATES AT THE VERTICAL IONIZATION POINTS Following the ionization, the reaction point was vertically shifted
from the ground to ionization state. Hereafter, the vertical ionized states of (Bz)n and (Bz)n-H2O are expressed as [(Bz)n+]ver and [(Bz)n+-H2O]ver, (_n_ = 2 and 3), respectively, where
[X+]ver indicates a radical cation of X at the vertical ionization point from its parent neutral species X. In [(Bz)2+]ver, the NPA molecular charges on (Bz)d and (Bz)a were calculated to be
+0.659 and +0.341, respectively, indicating that a positive charge was asymmetrically distributed on the benzene dimer cation (Table 1): the value of the charge on (Bz)d was about two times
larger than that of (Bz)a. In [(Bz)2-H2O+]ver, the charges changed to +0.818 in (Bz)d, +0.177 in (Bz)a, and +0.005 in H2O. The positive charge on (Bz)d increased because of its interaction
with H2O, whereas the positive charge on (Bz)a was decreased by H2O. The magnitude of the asymmetry in the charge distribution on (Bz)2+ was enhanced by H2O. The spatial distributions of the
spin density of [(Bz)2+]ver and [(Bz)2-H2O+]ver are illustrated in Fig. S1. In [(Bz)2+]ver, the calculated spin densities on (Bz)d and (Bz)a were 0.661 and 0.339, respectively, indicating
that the distribution of the unpaired electron on (Bz)d was about twice as large as that of (Bz)a (Table 1). In [(Bz)2+-H2O]ver, the spin densities were 0.821 on (Bz)d, 0.180 on (Bz)a, and
−0.001 on H2O, indicating that the positive charge and spin density were pushed out from (Bz)d to (Bz)a by H2O because of the positive charge of the proton of H2O in [(Bz)2-H2O+]ver. Thus,
the distribution of electrons was strongly influenced by H2O in the cation system, and the asymmetry of the spin density distribution was enhanced by H2O. The NPA charge and spin densities
of the trimer system (_n_ = 3) are given in Table 2. In the vertical ionized states of (Bz)3, [(Bz)3+]ver, the NPA molecular charges on (Bz)d, (Bz1)a, and (Bz2)a were calculated to be
+0.679, +0.161, and +0.161, respectively, indicating that a positive charge was symmetrically distributed on the benzene trimer cation; the value of the charge on (Bz)d was about four times
larger than that of (Bz)a. In the vertical ionized states of (Bz)3-H2O, [(Bz)2-H2O+]ver, the charges were +0.717 in (Bz)d, +0.043 in (Bz1)a, +0.237 in (Bz2)a, and +0.003 in H2O. The
magnitudes of the positive charges on (Bz)d and (Bz2)a were enhanced by interaction with H2O, whereas the positive charge on (Bz1)a was decreased by H2O. In [(Bz)3+]ver, the spin densities
on (Bz)d, (Bz1)a, and (Bz2)a were 0.684, 0.158, and 0.158, respectively, indicating that the distribution of the unpaired electron on (Bz)d was larger than those of (Bz1)a and (Bz2)a. In
[(Bz)2+-H2O]ver, the spin densities were 0.721 on (Bz)d, 0.044 on (Bz1)a, 0.235 on (Bz2)a, and −0.001 on H2O, indicating that the spin density was pushed out from (Bz)d to (Bz)a by H2O. This
result suggested that H2O created asymmetry of the electronic structure in the radical cation state. Π-STACKING FORMATION IN (BZ)2-H2O+ POTENTIAL ENERGY The time evolution of the potential
energy of (Bz)2+-H2O, following the ionization of the parent neutral complex, is given in Fig. 2 (top). Also, the snapshots of (Bz)2+-H2O are shown in Fig. 2 (bottom). In this trajectory
calculation, the optimized structure obtained at the CAM-B3LYP/6-311++G(d,p) level was used as the initial structure (time zero), and direct AIMD calculations were carried out at the
CAM-B3LYP/6-31 G(d) level. The zero level on the vertical axis corresponds to the total energy of [(Bz)2+-H2O]ver at time zero. The reaction dynamics of (Bz)2+-H2O could be classified by six
regions as follows: region A: initial structural change, region B: collision of (Bz)d to (Bz)a and rotation of H2O, region C: rebound of (Bz)d from (Bz)a, region D: rotation of (Bz)d on
(Bz)a, region E: C-C bond formation, and region F: complete of π-stacking. After the ionization of (Bz)2-H2O (point a), the potential energy decreased suddenly to −2.0 kcal/mol within 5 fs.
This energy decrease was caused by an internal structural deformation of (Bz)2+ after hole capture (region A). Then, the energy gradually decreased because of the changes in the structural
conformation between (Bz)d and (Bz)a and the rotation of H2O on (Bz)2+. (Bz)d gradually approached (Bz)a, and the collision of (Bz)d to (Bz)a occurred at 212 fs (point b in region B). In
addition, the rotation of H2O occurred as a result of the repulsive interactions between the positive charges of the proton (H2O) and Bz+. The stabilization energy of the rotation of H2O was
−4.0 kcal/mol, while the energy generated by collision of Bz+ with Bz was −2.0 kcal/mol. The internal structural deformation was −2.0 kcal/mol (0–5 fs). The total potential energy was −8.0
kcal/mol at point b. Moreover, the potential energy increased to −2.0 kcal/mol due to the rebound of (Bz)d from (Bz)a (region C). In conjunction with the rebound, the rotation of (Bz)d on
(Bz)a occurred gradually, and the potential energy decreased again together with the vibration (region D). The snapshot at point C (417 fs) indicated that the structure of (Bz)2+ deformed
gradually from a T-shape to the π-stacking form. In region E, the potential energy further decreased because a C-C bond was gradually formed between (Bz)d and (Bz)a. At point d (594 fs in
region F), the π-stacking formation was fully complete (final stage, region F). The potential energy reached a minimum point at 594 fs (point d). Thus, the time of π-stacking formation was
calculated to be 594 fs in this trajectory. SNAPSHOTS Figure 2 (bottom) shows snapshots of (Bz)2+-H2O following the ionization of (Bz)2-H2O. The intermolecular distances between the benzene
molecules were 4.151 Å for R1 and 3.722 Å for (Bz)a-H2O (r1). After ionization, the rotation of H2O on (Bz)2+ occurred, and (Bz)d gradually approached (Bz)a and collided at 212 fs (point b).
After the collision, (Bz)d rotated on (Bz)a. The arrows schematically indicate the direction of rotation. At 594 fs, the intermolecular distance was R1 = 2.693 Å and the π-stacking
formation was complete. It should be emphasized here that the H2O molecule showed a specific behavior after the ionization. The dipole of H2O oriented toward the center of mass of (Bz)a at
time zero (point a). After the ionization, H2O rotated rapidly on (Bz)a, and the oxygen atom of H2O interacted with (Bz)a (regions A and B). After rotation, the oxygen atom of H2O oriented
toward (Bz)a (regions B-D). In region D, a CH-OH2 hydrogen bond was formed between (Bz)a and H2O. In the final stage (point d in region F), the hydrogen bond and π-stacking formation were
complete. Π-STACKING FORMATION IN (BZ)2 + WITHOUT H2O In the case of (Bz)2+-H2O, the π-stacking formation was complete at 594 fs. In this section, to elucidate the effects of H2O on the time
of π-stacking formation, a similar direct AIMD calculation was carried out for the non-water system, (Bz)2. Figure 3 shows the potential energy and snapshots of (Bz)2+ following the
ionization of (Bz)2. After the ionization, (Bz)d gradually approached (Bz)a and collided with (Bz)a at 171 fs. The T-shaped structure was maintained at point c. (Bz)d rebounded from (Bz)a
(496 fs, point c) and a second collision occurred at 663 fs (point d). After the second collision, both (Bz)d and (Bz)a rotated (791 fs, point e), and π-stacking was complete at 920 fs
(point f). The time of π-stacking formation was 920 fs in this trajectory. The timescale for (Bz)2+ was about 300 fs longer than that of (Bz)2+-H2O, suggesting that one water molecule
largely accelerated the π-stacking formation of (Bz)2. EFFECTS OF WATER MOLECULE ON TIME OF Π-STACKING FORMATION In previous sections, it has been discussed that a water molecule accelerated
the π-stacking formation in a benzene dimer cation. In this section, the reasons for the acceleration of the formation time by H2O are discussed based on theoretical results. Figure 4 shows
the potential energies and intermolecular distances between the benzene rings plotted as a function of time for (Bz)2+ without H2O (denoted as _no-H_2_O_) and (Bz)2+-H2O (denoted as
_H_2_O_). At 200 fs, the energy minima corresponding to the collision state were found in both systems. However, the values of energies were large different: −8.0 kcal/mol (_H_2_O_) and −4.0
kcal/mol (_no-H_2_O_). This difference was caused by the rotation of H2O on (Bz)2+. The rotation significantly stabilized the energy of the collision state of (Bz)d-(Bz)a+ because of the
electrostatic effect. After the collision, (Bz)d rebounded from (Bz)a. In the case of _H_2_O_, the distance of (Bz)d from (Bz)a was calculated to be 3.5 Å at the turning point (400 fs),
whereas the distance was 4.1 Å in _no-H_2_O_ (480 fs). The distance at the turning point in _no-H_2_O_ was 0.6 Å longer than that of _H_2_O_. In addition to the longer distance, the time in
the bound state for _no-H_2_O_ was significantly longer than that of _H_2_O_ (200 vs. 350 fs). Also, the plateau of the potential energy caused by the rotation of (Bz)d on (Bz)a was found at
750–800 fs for _no-H_2_O_. In contrast, the rotation barrier disappeared in _H_2_O_. Thus, the effects of H2O on the reaction dynamics can be summarized as follows: (1) The excess energy
caused by the rotation of H2O on (Bz)2+ contributed to acceleration of π-stacking formation, with the water molecule stabilizing the potential curve as a whole. (2) H2O could decrease the
lifetimes of collision and rebound states as the oxygen atom of H2O attracts (Bz)d+ through electrostatic interactions. (3) The rotation barrier between Bz-Bz was also diminished by H2O.
EFFECT OF INITIAL STRUCTURE ON TIME OF Π-STACKING FORMATION In the previous sections, it was shown that the water molecule accelerated the π-stacking formation in (Bz)2+. However, the
results were obtained from only one trajectory for the (Bz)2 and (Bz)2-H2O systems. In this section, the effect of the initial geometry on the time of π-stacking formation was investigated
in detail. Six levels of theory were examined in the geometry optimizations. The geometry optimizations were carried out using A: 6-311++G(2d,p), B: 6-311++G(2d,2p), C: 6-311++G(2df,2p), D:
6-311++G(2df,2pd), E: 6-311++G(3df,2pd), and F: 6-311++G(3df,3pd) basis sets and the results are presented in Fig. 5. In the case of _no-H_2_O_, the time of π-stacking formation was
calculated to be 780–907 fs, while the time was distributed in the 432–621 fs range in _H_2_O_. The wide distribution in H2O occurred because the position of H2O on (Bz)2 was slightly
dependent on the basis sets used in the geometry optimizations. All levels of theory indicated that H2O significantly accelerated the time of π-stacking formation. The initial geometries
were also generated by thermal activation at 10 K. First, the geometries of the neutral dimer (Bz)2 and complex (Bz)2-H2O were fully optimized at the CAM-B3LYP/6-311++G(d,p) level. From
these geometries, direct AIMD calculations of the neutral systems were carried out at constant temperature conditions at the CAM-B3LYP/6-311++G(d,p) level. The structures of (Bz)2 and
(Bz)2-H2O fluctuated slightly around the equilibrium structures under this thermal condition (10 K). Twelve geometries were selected from each direct AIMD calculation of the cation state,
which were performed at the CAM-B3LYP/6-31G(d) level. The results are given in Table 3 (10 K distribution). The times of π-stacking formation were calculated to be 457 fs (_H_2_O_) and 691
fs (_no-H_2_O_). From thermal sampling calculations, it was also found that H2O accelerated the π-stacking formation. To check the methodology dependence on the accelerating effect of H2O,
direct AIMD calculations were carried out using a cc-pVDZ basis set with the CAM-B3LYP/ 6-311++G(d,p) optimized geometry. The results are given in Table 3 (cam-cc-pVDZ). The times of
π-stacking formation were calculated to be 481 fs (_H_2_O_) and 1068 fs (_no-H_2_O_). From all the calculations, it was concluded that H2O accelerates the π-stacking formation in the benzene
dimer cation. Π-STACKING FORMATION IN (BZ)3-H2O+ SNAPSHOTS Figure 6 shows the snapshots of (Bz)3+-H2O following the ionization of (Bz)3-H2O. The optimized structure obtained at the
CAM-B3LYP/6-311++G(d,p) level was used as the initial structure in the direct AIMD calculation (0 fs, point a). The intermolecular distances between the benzene molecules were 4.145 Å for R1
and 4.141 Å for R2, and the (Bz1)a-H2O distance (r1) was 3.612 Å. After ionization, both benzene molecules in the wing sites of (Bz1)a and (Bz2)a gradually approached (Bz)d and collided at
298 fs (point b). After the collision, the three benzene rings rotated with respect to each other. The arrows schematically indicate the direction of rotation. At 463 fs, the intermolecular
distances were R1 = 3.375 Å and R2 = 3.413 Å, and the structure of (Bz)3+ gradually approached the π-stacking form. At 566 fs (point d), the π-stacking form was complete. The behavior of H2O
around (Bz)3+ was very similar to that of _n_ = 2 (benzene dimer cation). The snapshots of (Bz)3+ without H2O following the ionization of (Bz)3 are shown in Fig. S2. The reaction dynamics
of (Bz)3+ were very similar to those of the benzene dimer cation (Fig. 3). The time of π-stacking formation was calculated to be 1155 fs in (Bz)3+ (_no-H_2_O_). POTENTIAL ENERGIES The time
evolution curves of the potential energy of (Bz)3+-H2O and (Bz)3+, following the ionization of the parent neutral complex, are shown in Fig. 7. After the ionization of (Bz)2-H2O, the
potential energy decreased gradually, and the collision of (Bz1)a and (Bz2)a to (Bz)d occurred at 298 fs. After the collision, the three benzene rings rotated with respect to each other, and
π-stacking formation was complete at 566 fs. In the case of (Bz)3+, the π-stacking formation was complete at 1155 fs. Thus, the H2O molecule significantly accelerated the π-stacking
formation in the benzene trimer cation as well as in the benzene dimer. The other levels of theory, i.e., the CAM-B3LYP/6-311++G(2d,p) and 6-311++G(2d,2p) optimized structures, gave similar
results (Table 4). DISCUSSION REACTION MODEL Based on the results derived from the calculations presented above, a model was proposed for the effect of H2O on the timescale of the ON-OFF
switching element composed of a benzene cluster. Figure 8 shows a schematic illustration of the proposed model. In the neutral state (upper figure), the C-H-π interaction is dominant between
the benzene molecules, and the benzene cluster forms a non-stacking T-shape structure. After hole capture, the structure drastically changes from non-stacking to π-stacking forms. The hole
can easily move along the stacked benzene rings. When the π-stacking form captures an electron, the structure spontaneously returns to the non-stacking form. If a water molecule interacts
with a benzene molecule, the stacking rate is significantly accelerated because of the asymmetry of the electronic structure in the benzene rings. Nowadays, the surface functionalized
graphenes have been designed and synthesized as molecular devices30,31. The present model would be applied to these molecular systems in near future. Also, the present investigation has open
a field of the small cluster electronic devices. CONCLUDING REMARKS In the present calculations, several assumptions were employed in the ab initio calculations. First, the CAM-B3LYP
functional was used in the direct AIMD calculation, and subsequently, the π-stacking reactions were discussed. To check the functional dependence on reaction time, several functionals were
examined in the direct AIMD calculations. The results are given in Table 5 (benzene dimer, _n_ = 2) and 6 (benzene trimer, _n_ = 3). The direct AIMD calculations with the APFD functional
showed that the time of π-stacking was 358 fs (_H_2_O_) and 883 fs (_no-H_2_O_) for _n_ = 2. In the case of _n_ = 3, these times were calculated to be 434 fs (_H_2_O_) and 1005 fs
(_no-H_2_O_). All results shown in Tables 5 and 6 suggest that H2O accelerates the π-stacking formation in both benzene dimer and trimer cation. As for the other factors, the position of H2O
around neutral (Bz)n (_n_ = 2 and 3), methanol-(Bz)n (instead of H2O), and zero-point vibration were also examined (supporting information). CONCLUSION The calculations presented herein
revealed that a H2O molecule accelerates the time of π-stacking formation in a benzene molecular system. The times of stacking formation in the benzene dimer (_n_ = 2) and trimer (_n_ = 3)
cations were calculated to be 594 fs (_H_2_O_) and 922 fs (_no-H_2_O_), and 566 fs (_H_2_O_) and 1155 fs (_no-H_2_O_), respectively. Thus, H2O showed a positive effect in benzene-based
molecular electronics. This tendency was not dependent on the level of theory used for calculations. The acceleration primarily originated from the re-orientation of H2O on benzene cluster
cation following the hole capture. While previous studies have shown that H2O suppresses the conductance and destroys the circuit of electron transport in molecular electronics (i.e., has
negative effects)14,15,16,17,18,19,20, the present study demonstrated that H2O produces positive effects in benzene-based molecular electronics. REFERENCES * Balliou, A. _et al_.
Size-dependent single electron transfer and semi-metal-to-insulator transitions in molecular metal oxide electronics. _Nanotechnology_ 29, 275204 (2018). Article Google Scholar * Xiang,
D., Wang, X. L., Jia, C. C., Lee, T. & Guo, X. F. Molecular-scale electronics: from concept to function. _Chem. Rev._ 116, 4318–4440 (2016). Article CAS Google Scholar * Sun, M.,
Zhang, Z., Kim, Z. H., Zheng, H. & Xu, H. Plasmonic scissors for molecular design. _Chem. Eur. J._ 19, 14958–14962 (2013). Article CAS Google Scholar * Xiang, D., Jeong, H., Lee, T.
& Mayer, D. Mechanically controllable break junctions for molecular electronics. _Adv. Mater._ 25, 4845–4867 (2013). Article CAS Google Scholar * Cui, X. D. _et al_. Reproducible
measurement of single-molecule conductivity. _Science_ 294, 571–574 (2001). Article ADS CAS Google Scholar * Wang, Q. _et al_. Single-atom switches and single-atom gaps using stretched
metal nanowires. _ACS Nano_ 10, 9695–9702 (2016). Article CAS Google Scholar * Taniguchi, M., Tsutsui, M., Shoji, K., Fujiwara, H. & Kawai, T. Single-molecule junctions with strong
molecule-electrode coupling. _J. Am. Chem. Soc._ 131, 14146–14147 (2009). Article CAS Google Scholar * Bian, B. _et al_. First-principles study on photoswitching behavior and negative
differential resistance in single molecule junction. _Comput. Theor. Chem._ 1115, 185–189 (2017). Article CAS Google Scholar * Bian, B. _et al_. Effect of the substitution of F on the
photoswitching behavior in single molecular device. _Phys. Lett._ 381, 2748–2753 (2017). Article CAS Google Scholar * Brandbyge, M. _et al_. Density-functional method for nonequilibrium
electron transport. _Phys. Rev. B_ 65, 165401 (2002). Article ADS Google Scholar * Fan, Z.-Q. & Chen, K.-Q. Stable Two-Dimensional Conductance Switch of Polyaniline Molecule
Connecting to Graphene Nanoribbons. _Sci. Rep._ 4, 5976 (2014). Article CAS Google Scholar * Guo, X. F. _et al_. Covalently bridging gaps in single-walled carbon nanotubes with conducting
molecules. _Science_ 311, 356–359 (2006). Article ADS CAS Google Scholar * Yang, J. J., Han, X. X., Yuan, P. P., Bian, B. A. & Wang, Y. X. Effect of different substitution position
on the switching behavior in single-molecule device with carbon nanotube electrodes. _Chem. Phys._ 500, 74–79 (2018). Article CAS Google Scholar * Li, Z. L. _et al_. Effect of H2O
Adsorption on Negative Differential Conductance Behavior of Single Molecular Junction. _Sci. Rep._ 7, 4195 (2017). Article ADS Google Scholar * Tachikawa, H. & Kawabata, H. A density
functional theory study on the degradation mechanism of thin film of organic semiconductor by water molecules. _Thin Solid Films_ 516, 3287–3293 (2008). Article ADS CAS Google Scholar *
Tachikawa, H. Effects of micro-solvation on the reaction dynamics of biphenyl cations following hole capture. _Chem. PHys._ 490, 12–18 (2017). Article CAS Google Scholar * Long, D. P. _et
al_. Effects of hydration on molecular junction transport. _Nature Mater._ 5, 901–908 (2006). Article ADS CAS Google Scholar * Li, X. L. _et al_. Thermally activated electron transport
in single redox molecules. _J. Am. Chem. Soc._ 129, 11535–11542 (2007). Article CAS Google Scholar * Na, J. S., Ayres, J., Chandra, K. L., Gorman, C. B. & Parsons, G. N. Real-time
conductivity analysis through single-molecule electrical junctions. _Nanotechnology_ 18, 424001 (2007). Article Google Scholar * Cao, H., Jiang, J., Ma, J. & Luo, Y.
Temperature-dependent statistical behavior of single molecular conductance in aqueous solution. _J. Am. Chem. Soc._ 130, 6674–6675 (2008). Article CAS Google Scholar * Tachikawa, H.
Jahn-Teller Effect of the Benzene Radical Cation: A Direct ab Initio Molecular Dynamics Study. _J. Phys. Chem. A_ 122, 4121–4129 (2018). Article CAS Google Scholar * Tachikawa, H. Effects
of Zero Point Vibration on the Reaction Dynamics of Water Dimer Cations following Ionization. _J. Comput. Chem._ 38, 1503–1508 (2017). Article CAS Google Scholar * Tachikawa, H. Reaction
Dynamics Following Ionization of Ammonia Dimer Adsorbed on Ice Surface. _J. Phys. Chem. A_ 120, 7301–7310 (2016). Article CAS Google Scholar * Tachikawa, H., Miyazawa, Y. & Iura, R.
Timescale of pi-Stacking Formation in a Benzene Trimer Cation Formed by Ionization of the Parent Neutral Trimer: A Direct Ab Initio Molecular Dynamics Study. _Chem. Slect._ 3, 1113–1119
(2018). CAS Google Scholar * Tachikawa, H. Direct ab initio molecular dynamics (MD) study of the ionization on a benzene dimer. _RSC Adv._ 2, 6897–6904 (2012). Article CAS Google Scholar
* Tachikawa H. Double pi-pi stacking dynamics of benzene trimer cation: direct ab initio molecular dynamics (AIMD) study, _Theor. Chem. Acc_. 132, UNSP 1374 (2013). * Frisch M. J. _et al_.
_Gaussian09_, Rev. D.01, Gaussian Inc., Wallingford, CT, (2009). * Tachikawa, H. Electronic states of hydrogen atom trapped in diamond lattice. _Chem. Phys. Lett._ 513, 94–98 (2011).
Article ADS CAS Google Scholar * Tachikawa, H. & Shimizu, A. Diffusion Dynamics of the Li Atom on Amorphous Carbon: A Direct Molecular Orbital-Molecular Dynamics Study. _J. Phys.
Chem. B_, 110, 20445–20450 (2006). Article CAS Google Scholar * Hongwu, C., Chun, L. & Liangti, Q. Solution electrochemical approach to functionalized graphene: History, progress and
challenges. _Carbon_ 140, 41–56 (2018). Article Google Scholar * Tachikawa, H. & Kawabata, H. Electronic states of aryl radical functionalized graphenes: Density functional theory
study. _Jpn. J. Appl. Phys._ 55, 06GK05 (2016). Article Google Scholar Download references ACKNOWLEDGEMENTS The author (H.T.) acknowledges partial support from JSPS KAKENHI Grant Numbers
18K05021 and 17H03292. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Division of Applied Chemistry, Graduate School of Engineering, Hokkaido University, Sapporo, 060-8628, Japan Hiroto
Tachikawa, Ryoshu Iura & Hiroshi Kawabata Authors * Hiroto Tachikawa View author publications You can also search for this author inPubMed Google Scholar * Ryoshu Iura View author
publications You can also search for this author inPubMed Google Scholar * Hiroshi Kawabata View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
H.T. proposed the idea of this research. R.I. and H.T. carried out the ab initio and direct AIMD calculations, the characterizations and theoretical analysis. H.K. carried out the energy
calculations of potential energy curves and zero point energies of systems. H.T. wrote the manuscript, and supervised and finalized the project. CORRESPONDING AUTHOR Correspondence to Hiroto
Tachikawa. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPORTING_INFORMATION RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a
Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are
included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Tachikawa, H., Iura, R. & Kawabata, H. Water-accelerated
π-Stacking Reaction in Benzene Cluster Cation. _Sci Rep_ 9, 2377 (2019). https://doi.org/10.1038/s41598-019-39319-7 Download citation * Received: 05 October 2018 * Accepted: 21 January 2019
* Published: 20 February 2019 * DOI: https://doi.org/10.1038/s41598-019-39319-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable
link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Javascript support required...
Dinner and a movie: ‘beetlejuice’ | members only accessWhat follows is a loose lesson on villains and heroes. Who is the real hero here, anyway? Is it the crusty Beetlejuice, ...
Open access | npj breast cancer_npj Breast Cancer_ makes all content freely available to all researchers worldwide, ensuring maximum dissemination of c...
'this little light of mine' shines on, a timeless tool of resistance_This story is part of American Anthem, a yearlong series on songs that rouse, unite, celebrate and call to action. Find...
Cut senior year in half (opinion)With apologies to Winston Churchill, rarely have _so many done so little for so long_ as college-bound seniors during th...
Latests News
Water-accelerated π-stacking reaction in benzene cluster cationABSTRACT Single molecule electron devices (SMEDs) have been widely studied through both experiments and theoretical calc...
Imf slashes world growth forecasts again on brexit curveballThe U.K's vote to leave the European Union (EU) has pushed the International Monetary Fund (IMF) to cut its world g...
Toyota close to $1 billion deal to settle probe: wsjToyota Motor is close to a deal to pay $1 billion to settle a U.S. criminal investigation into how it disclosed customer...
Former newcastle owner mike ashley sees net worth tumble by £679mFormer Newcastle United owner Mike Ashley has seen his wealth drop by £679 million, according to the latest edition of T...
Center-environment feature models for materials image segmentation based on machine learningABSTRACT Materials properties depend not only on their compositions but also their microstructures under various process...