Integrative determination of atomic structure of mutant huntingtin exon 1 fibrils implicated in huntington disease
Integrative determination of atomic structure of mutant huntingtin exon 1 fibrils implicated in huntington disease"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Neurodegeneration in Huntington’s disease (HD) is accompanied by the aggregation of fragments of the mutant huntingtin protein, a biomarker of disease progression. A particular
pathogenic role has been attributed to the aggregation-prone huntingtin exon 1 (HTTex1), generated by aberrant splicing or proteolysis, and containing the expanded polyglutamine (polyQ)
segment. Unlike amyloid fibrils from Parkinson’s and Alzheimer’s diseases, the atomic-level structure of HTTex1 fibrils has remained unknown, limiting diagnostic and treatment efforts. We
present and analyze the structure of fibrils formed by polyQ peptides and polyQ-expanded HTTex1 in vitro. Atomic-resolution perspectives are enabled by an integrative analysis and
unrestrained all-atom molecular dynamics (MD) simulations incorporating experimental data from electron microscopy (EM), solid-state NMR, and other techniques. Alongside the use of prior
data, we report magic angle spinning NMR studies of glutamine residues of the polyQ fibril core and surface, distinguished via hydrogen-deuterium exchange (HDX). Our study provides a
molecular understanding of the structure of the core as well as surface of aggregated HTTex1, including the fuzzy coat and polyQ–water interface. The obtained data are discussed in context
of their implications for understanding the detection of such aggregates (diagnostics) as well as known biological properties of the fibrils. SIMILAR CONTENT BEING VIEWED BY OTHERS THE
STRUCTURE OF PATHOGENIC HUNTINGTIN EXON 1 DEFINES THE BASES OF ITS AGGREGATION PROPENSITY Article 02 March 2023 CRYO-ELECTRON TOMOGRAPHY PROVIDES TOPOLOGICAL INSIGHTS INTO MUTANT HUNTINGTIN
EXON 1 AND POLYQ AGGREGATES Article Open access 08 July 2021 HUNTINGTIN FIBRILS WITH DIFFERENT TOXICITY, STRUCTURE, AND SEEDING POTENTIAL CAN BE INTERCONVERTED Article Open access 13 July
2021 INTRODUCTION Huntington’s disease (HD) is one of a family of incurable neurological genetic disorders resulting from an aberrant expansion of a CAG trinucleotide repeat in a
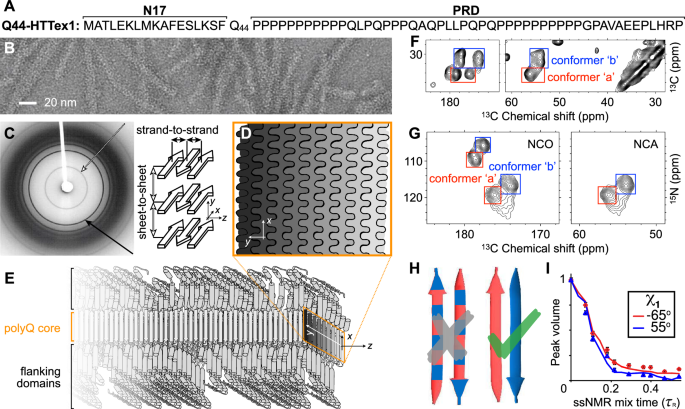
disease-specific gene1,2. In HD, the mutation impacts the huntingtin (HTT) protein, which ends up with an expanded polyglutamine (polyQ) tract in its first exon (HTTex1, Fig. 1A)3. In HD
patients and model animals, mutant HTT fragments matching HTTex1 form inclusions in brain areas affected by neurodegeneration. In cells, _μ_m-sized inclusions contain fibrillar protein
aggregates with widths of several nanometers, as seen by cryogenic electron tomography (cryo-ET)4,5,6. Fibrillar HTTex1 has all the common features of amyloid fibrils found in diseases such
as Parkinson’s and Alzheimer’s disorders: highly stable protein deposits built around extended intermolecular _β_-sheets forming a characteristic cross-_β_ architecture7,8,9. The protein
fibrils are of interest due to their potential involvement in pathogenic and neurotoxic processes—but also as biomarkers of disease onset and progression. Consequently, there is a need to
resolve their atomic structures to facilitate the design of inhibitors, modulators, and diagnostic tools. For instance, a better understanding of HTTex1 fibril structure is essential to the
development of ligands used to detect HTTex1 fibril formation in vivo10. Recent years have seen important progress in amyloid structure determination through application of cryogenic
electron microscopy (cryo-EM) and solid-state NMR spectroscopy (ssNMR)7,11,12. Breakthrough studies have produced structures of amyloid fibrils formed by tau, amyloid-_β_, and _α_-synuclein,
associated with Alzheimer’s, Parkinson’s, and other amyloid disorders12. The polyQ-based amyloid fibrils from HD and other CAG repeat disorders2, however, have proved more challenging, and
still lack truly atomic-level structures or structural models, limiting progress in mechanistic and diagnostic research. The challenges in studying these proteins stem in part from their
atypical amyloidogenic motif, formed by repetitive sequences. The aggregation propensity of polyQ proteins correlates strongly with their polyQ tract length, as determined by the inherited
mutation, which inversely correlates with the age of onset2,13. In their native state, polyQ segments are typically considered to lack a stable secondary structure, forming an intrinsically
disordered region. This is also true for the polyQ-containing segment of HTT (HTTex1), which is invisible in cryo-EM of the unaggregated full-length human HTT protein14,15. However, in HD,
N-terminal fragments of polyQ-expanded HTT are generated from protease activity and aberrant splicing, enabling their subsequent aggregation into fibrils and ultimately cellular
inclusions16,17,18. This has lead to considerable interest in studies of HTTex1 aggregates as an important mechanistic factor in HD pathogenic mechanisms. In in vitro structural studies,
HTTex1 fibrils (Fig. 1B) featured a highly ordered amyloid core, formed by the polyQ segment, surrounded by flexible non-amyloid flanking regions6,19,20,21,22,23. Prior fiber X-ray
diffraction studies showed the core itself to comprise anti-parallel _β_-sheets in a cross-_β_ pattern (Fig. 1C)24,25,26,27. These experiments spurred proposals of various possible fibril
models24,25,26,27. However, such models were typically presented as illustrative rather than an atomic-level analysis, due to the lack of availability of sufficient experimental information.
Moreover, all early X-ray-based models proved inconsistent with later work8,28,29,30 that revealed important new structural data via combinations of cryo-EM, ssNMR, and other
techniques8,19,20,21,22,30,31,32,33,34,35,36. The current study builds on valuable data from various prior experimental studies, and provides additional (NMR) experimental data, to perform
an integrative structural analysis of polyQ amyloid and HTTex1 fibrils. A few recent studies are of particular interest. Firstly, an integration of ssNMR, X-ray, and EM measurements
permitted the manual assembly of a schematic fibril architecture in which the polyQ segments form a block-like fibril core (Fig. 1D–E)8,20,28,29. Subsequently, a cryo-EM study of HTTex1
fibrils23 produced a medium-resolution density map, which—although it lacked the detail for a de-novo atomistic structure—was interpreted to be consistent with the abovementioned model
architecture: the amyloid core has a block-like structure assembled from multiple layers of tightly packed _β_-sheets (as schematically shown in Fig. 1D–E). Notably, this type of
architecture is distinct from more typical amyloid fibrils (Supplementary Fig. 1). The apparent success of such a unified (but as-yet qualitative) model underlines the need for a modern
integrative-structural-biology approach37 to fully describe these fibrils that have much in common with other amyloids, but also a number of truly unique features. Here we employ rigorous
physics-based molecular modeling to integrate the collective experimental knowledge into atomic-resolution polyQ and HTTex1 fibril structures that are fully consistent with all the key
experimental data, including sheet-to-sheet distances from fiber X-ray diffraction, fiber dimensions from EM, and structural constraints from ssNMR. We combine previously reported
experimental data with additional experimental measurements, with the latter in particular examining the fibril core and surface features that were previously not observed. The obtained
structures are analyzed in the context of known structural features of HTTex1 amyloid fibrils, allowing a detailed explanation of their conformational and spectroscopic characteristics.
Moreover, based on the disposition and accessibility of key residues and segments, the molecular architecture of these HD-related fibrils explains biological properties, such as the
(in)ability for post-aggregation polyubiquitination and degradation. RESULTS AND DISCUSSION ARCHITECTURE OF THE INTERNAL POLYQ FIBRIL CORE We started the integrative structure determination
of HTTex1 fibril from its polyQ amyloid core. For this, we first review previously reported experimental data that inform our modeling approach. Most experimental studies, in particular
recent ssNMR studies (Supplementary Fig. 1A), argue for a shared molecular architecture common to polyQ-containing peptide and protein fibrils36,38. Various techniques show that polyQ
amyloid features antiparallel _β_-sheets (Fig. 1E)26,27,30,33,39, in contrast to the parallel in-register fiber architecture of many other amyloid fibers11,40. Another distinct feature of
polyQ amyloid is that it forms long _β_-strands with few turns (Fig. 1F; Supplementary Fig. 1B)8,23,27,29,41, unlike the majority of other amyloid structures that have short _β_-strands
connected by turns or loops11. The polyQ fiber core is multiple nanometers wide, devoid of water molecules, and forms a block-like structure featuring a stacking of less than ten _β_-sheets
(Fig. 1D)23,27,28,29. Its cross-section must contain polyQ segments from multiple protein monomers (schematically illustrated in Fig. 1D–E)41. A large proportion of the Gln residues is
buried, far away from the solvent, in a repetitive and pseudo-symmetric context. This ordered and repetitive nature is demonstrated by high-quality ssNMR spectra, with few and relatively
narrow peaks (Fig. 1F–G; Supplementary Fig. 1)8,20,22,28,29,32,41. These ssNMR studies consistently show a characteristic spectroscopic fingerprint that identifies two dominant Gln residue
conformations—see the two sets of peaks from so-called “a” and “b” conformers (red and blue boxes) present at roughly equal intensities in the 2D ssNMR spectra of Q44-HTTex1 fibrils (Fig.
1F–G; Supplementary Fig. 1). The two Gln conformers reflect two types of _β_-strand structures that co-assemble into a single antiparallel _β_-sheet (Fig. 1H; more below)8,22,29. Integrating
these known experimental data, we concluded that the internal polyQ core structure can be captured by an infinitely-repeating eight-Gln unit that features antiparallel _β_-sheets (Fig. 2A).
This construction has translational symmetry and lacks the twisting seen in many amyloid fibrils, since EM analyses of our and others’ HTTex1 fibrils lack strong signs of twisting23. The
repeating unit cell (Fig. 2A) fulfills several a priori requirements: First, its peptide chain segments are two residues long, the minimum needed to capture the odd/even side-chain
alternation in a _β_-strand. Given that ssNMR shows only two ssNMR-detected conformers, in long strands (Supplementary Fig. 1; see refs. 8,22,29), two residues should be sufficient to
capture the structural variation within this amyloid core. Second, the unit cell contains two _β_-strands (that can differ in conformation), the minimum needed to describe an antiparallel
_β_-sheet. This contrasts with parallel in-register _β_-sheets that could be represented with a single repeating _β_-strand. Third, the unit cell contains a stacking of two _β_-sheets, to
permit the probing of distinct sheet–sheet dispositions. Fourth, certain ranges of side-chain dihedral angles (_χ_1, _χ_2, _χ_3) are required in order to fulfill a crucial feature of the
interdigitating polyQ amyloid:8,26 the side-chain–side-chain hydrogen bonding between the strands (see Fig. 2A and Supplementary Fig. 2). Thus, the _χ_2 dihedral must be close to 180∘, as
also known from ssNMR and Raman experiments8,42. This minimal model (Fig. 2A) fulfills the above-noted experimental constraints, and still yields a library of roughly 1 000 distinct
architectures (see Methods for details). A key additional consideration is that ssNMR has unambiguously shown sequential residues within each _β_-strand to have the same backbone
conformations (based on identical chemical shifts; Supplementary Fig. 1B)8,22,36. In summary, the “a" and “b" conformers strictly occupy distinct strands, within which uniform
backbone and _χ_1 dihedral angles are found (Fig. 1H). Moreover, these two distinct _β_-strand types differ in their _ψ_ and _χ_1 dihedral angles (Fig. 1I; Supplementary Fig. 1D, E)8. Thus,
these additional constraints can be applied to the initial library of architectures. After applying this additional filtering, 30 distinct unit cell architectures still remained possible
(Supplementary Fig. 3). To investigate the structural stability of these 30 models, we carried out all-atom MD simulations in a fully periodic infinite-core setting; see Methods for details.
Only two of the candidate models proved stable throughout the simulations (Fig. 2B and Supplementary Fig. 4). ZIPPERS AND LADDERS WITHIN THE POLYQ CORE Notably, these two stable 3D lattices
(denoted _M1_ and _M2_) capture, but also refine, known features of polyQ amyloid structure. Experimental constraints offered by previously reported ssNMR dihedral angle measurements are
open to more than one interpretation8,43. Previously, this inherent ambiguity yielded qualitative models useful for illustrative purposes. Here, we have rigorously narrowed down the viable
and physically plausible models, obtaining only two models that identify specific narrow regions within the equivocal dihedral angle space (Fig. 2D). Interestingly, unlike all the 28
unstable models, _M1_ and _M2_ display no Gln residues with _χ_3 ≈ 180∘ (Supplementary Fig. 6). In both models, the antiparallel _β_-sheet harbors strand-specific Gln structures occupying
the side chain rotamers known as pt20∘ and mt-30∘, as defined by ref. 44. These conformations enable hydrogen bonding interactions between the side chains of the Gln conformers, in addition
to typical strand-to-strand backbone hydrogen bonds (Fig. 2E). This hydrogen bonding pattern is reminiscent of glutamine (or asparagine) ladders found in other amyloids45,46. However, a key
difference is that here they occur in an antiparallel _β_-sheet, whereas most prior cases are parallel. A recent paper46 provided a detailed analysis of this type of ladder in HET-s fibrils,
noting the relevance of 1H shifts of the side chain amide nitrogens. Their hydrogen-bond strength has a noted effect on these 1H shifts47,48, resulting in a characteristic pattern for
glutamine side chains H-bonded in such ladders: a big difference between their HZ and HE shifts (nomenclature in Fig. 2F), with the former reflecting hydrogens involved in strong H-bonds
along the fiber axis. Thus, we performed 2D NMR to detect and assign these protons in the polyQ amyloid core (Fig. 2G, Supplementary Fig. 7). These spectra show the backbone N–H signals, as
well as peaks for the side chain NH2 groups of the “a” and “b” Gln conformers. Strikingly, these polyQ side chains feature widely separated HZ (~8.2 ppm) and HE (~5.5 ppm) 1H shifts. This
large HZ–HE shift difference actually exceeds that seen in the HET-s fibril46. This suggests a particularly strong H-bond interaction in polyQ amyloids, given that the 1H shift values for HZ
are connected to the H-bonding distance46,47,48. The resolution of the HET-s fibril structure does not permit a direct translation to an atomic distance, but analysis of Gln ladders in
other amyloids suggests side chain oxygen–nitrogen (heavy atom) distances of 2.7 to 2.9 Å49. Our stable structures for polyQ (Supplementary Fig. 8) show side-chain–side-chain H-bond
distances (for HZ H-bonds) that match such values. A notable feature of our models is that these observed distances are the same for the two “a”/“b” conformers. Residues in the HET-s fibril
core show substantial differences in their 1H shifts46, presumably due to differences in local structure and H-bonding distance. In contrast, the HZ and \({{\rm{H}}}_{\,\text{E}}\) 1H shifts
in the polyQ core are indistinguishable between the two conformers (Supplementary Fig. 7), consistent with the models’ indication that the corresponding hydrogen-bonding interactions are
alike for the two conformers in the polyQ core. Notably, these observations were not employed as restraints in our modeling setup. The close match between these simulated distances and the
provided NMR data on the Gln side chains (in the fibril core) offers experimental support for the validity of our modeling efforts. STRUCTURE OF POLYQ15 PEPTIDE FIBRILS The above 3D models
capture the internal core of polyQ protein fibrils, but lack the defined width and water-facing surface features of the real polypeptide fibrils. To model a proper experimental system, we
modified the infinite cores _M1_ and _M2_ into models of fibrils formed by the polyQ15 peptide (Fig. 3A). These widely studied8,24,27,50,51 peptides contain a 15-residue polyQ segment,
flanked by charged residues to enhance solubility. Experimental analysis8,29,52 by MAS NMR and X-ray diffraction has shown that atomic conformations in the polyQ15 fibril core match those of
the disease-relevant HTTex1 fibrils; e.g., the previously published8 ssNMR signals from Gln within the D2Q15K2 fibrils (Fig. 3A) and the Q44-HTTex1 fibrils (Fig. 1F) are indistinguishable.
Using the _M1_ and _M2_ models, we constructed D2Q15K2 fibrils comprising seven anti-parallel _β_-sheets, consistent with the 5.5–6.5-nm fibril width seen experimentally by TEM (Fig. 3A). We
then subjected both polyQ15 models to 1-_μ_s atomistic MD simulations in explicit water; for details, see Methods. Figure 3B shows the _M2_ D2Q15K2 fibril in cartoon representation. The
structure of the buried Gln residues within the core remained stable throughout the simulations (Supplementary Fig. 9). There is a high degree of agreement between the _χ_1, _χ_2, and _ψ_
dihedral angle distributions observed in MD simulations of our polyQ15 models and the ssNMR angle constraints previously reported, based on HCCH (_χ_1, _χ_2) and NCCN (_ψ_) experiments (Fig.
3C and Supplementary Fig. 10). Experiments8 have shown that the “a” and “b” ssNMR signals reflect different _χ_1 and _ψ_ values in the two Gln conformers, but a similar _χ_2 value. The
simulations of both polyQ15 models are consistent with these NMR data (Fig. 3C). The largest deviation affects the _M2_ model, in terms of its broader distribution of the conformer “a” _χ_1
angle, which in part lies outside the shaded region representing the (ambiguous) ssNMR constraint. Experimental studies have shown8 that the conformer “a” is more dynamic than conformer “b”,
with additional experimental evidence discussed below. These results are reminiscent of the increased heterogeneity of the “a” conformer in the MD simulations of both models (see also
Supplementary Fig. 9). This dynamic difference thus supports our MD results and dynamic averaging effects may help explain the noted (modest) deviation. Also, the sheet-to-sheet and
strand-to-strand distances within the models match well with repeat distances from X-ray diffraction (Supplementary Fig. 11). In sum, the MD simulations support the stability of these polyQ
amyloid core structures and recapitulate key data from multiple experimental techniques. SURFACE FEATURES OF POLYQ AMYLOID Our modeling reveals an important feature of polyQ protein or
peptide fibrils, which, so far, has not been seen or studied: the solvent-facing external structure of the polyQ amyloid core. The fiber surface mediates interactions with cellular
surroundings but also with any purposefully administered compounds, such as thioflavin-T or amyloid-specific tracers used in positron emission tomography (PET)10. In our models, the two
outermost _β_-sheets exposed to water (top and bottom sheets in Fig. 3B) allow us to compare the surface-exposed Gln residues versus the residues buried in the fibril core. Figure 4A
highlights the water-exposed Gln side-chains (green) in the polyQ15 fibril model. Figure 4 shows the _χ_1–_χ_2 and _χ_3–_χ_2 dihedral angle distributions of the core Gln residues (panel B,
gray) and compares them with the surface residues (panel C, green) for model _M1_ (see Supplementary Fig. 12 for _M2_). For the core residues, the rotamer states are similar to those
discussed above (pt20∘ and mt-30∘). For the surface residues, additional rotameric regions emerge, including Gln rotamers where _χ_2 deviates from 180∘, close to rotamer pm0∘44.
Interestingly, surface residues do not show fully free side-chain motion and retain some of the structural features that determine the molecular profile of the corrugated polyQ amyloid core
surface. This observation is highly relevant for efforts to design selective binders of the polyQ amyloid surface. Until now, few experimental data inform us about these polyQ–water
interfaces. A big challenge in understanding the surface-exposed Gln residues is that their NMR signals overlap with those of the core, which are more numerous and therefore dominate the
observed signals53. Indeed, we (the authors) had assumed the surface and core signals to be more similar than predicted by the MD results (Fig. 4). To evaluate this important feature, we
designed and performed experimental studies combining hydrogen–deuterium exchange (HDX) with advanced MAS NMR analysis. The N–H bonds within the fibril core are resistant to H/D exchange,
due to their dehydrated nature, stable hydrogen bonding, and lack of solvent access54. This feature allows one to differentiate and compare the NMR signatures of the core and surface of
polyQ protein fibrils. We produced Q44-HTTex1 fibrils that were aggregated either in regular (protonated) buffer or in deuterated buffer. In such fibrils, one expects the exchangeable amide
hydrogens to be either intact or exchanged for deuterium, respectively. By TEM, no deuteration-related effects were noted on the fibril morphology (Supplementary Fig. 15A, B). First,
1H-detected MAS NMR analysis of the fully protonated fibrils produced the 2D and 3D 1H–15N spectra shown in Fig. 5A and Supplementary Fig. 15D, featuring primarily signals from the rigid
polyQ core. Note that no strong signals are expected from the numerous HTTex1 proline residues, as they lack backbone amide protons. The observed signals match those expected for the
glutamine backbone amides (15N frequency 115–125 ppm) and the side chains (15N frequency near 100–115 ppm). Assignment of the polyQ core signals was based on abovementioned 2D correlation
experiments (Supplementary Fig. 7). Next, 2D MAS NMR was performed on the partly HDX-exchanged fibrils after exposure to protonated or deuterated buffer (Fig. 5B–C). One expects to either
observe only buried residues, or only exposed residues. These spectra reveal the protonated-core signals to match those of that we already assigned to polyQ amyloid (Supplementary Fig. 7).
However, the deuterated-core fibrils gave different peaks, which we attribute to the surface exposed glutamines (Fig. 5C). We will examine their distinct shifts below. As another key
indicator of surface-exposure, we also performed relaxation measurements (Supplementary Fig. 15G–J). We observed that the 15N backbone and side chains in the fibril core displayed the
relaxation characteristics of rigid residues. In contrast, the side chain 15N of the surface-exchangeable sites displayed faster relaxation (15N _T_1 and _T_1_ρ_). Exchangeable backbone 15N
(on the fibril surface) showed intermediate behavior that more closely resembled the polyQ core, in contrast to the side chains. Here it is important to note that the surface residues are
more mobile than their buried counterparts, but that their motion is nonetheless greatly restricted. First, these signals are detected via cross-polarization NMR that fails for flexible
residues (such as those in the PRD tail). Second, the relaxation properties are indicative of constrained motion, especially considering the relaxation properties of the observed backbone
signals. Thus, we experimentally observed solvent-exposed Gln residues accessible on the polyQ core surface. The chemical shifts of their backbone nitrogens are similar to those of the core
residues, suggesting a similarity to the amyloid core backbone structure. However, the side chain 15N and 1H shifts are very different from the core. Notably, whilst the characteristic side
chain HZ and HE shifts can be recognized quite clearly, they are much closer together than the already-discussed core residues. Thus, following the analysis discussed above, this implies the
absence of ordered Gln-ladders on the fibril surface. These experimental indicators of dynamics of the side chains on the surface, but more order of the backbones, are highly consistent
with the MD showing restricted dynamic disorder on the fibril core surface (Fig. 4C). STRUCTURE OF HD-RELEVANT HTT EXON 1 FIBRILS As already noted, protein inclusions seen in HD patients and
HD model animals incorporate the mutant HTTex1 protein fragment. Until now, no atomistic model of HTTex1 fibrils has been reported, although a diversity of schematic or cartoon-style models
has been published over the years. Here, we build on our above-presented polyQ amyloid core structures to construct an experiment-based molecular structure of Q44-HTTex1 fibrils. The
Q44-HTTex1 construct is used to model the disease-relevant HD protein in a variety of experimental studies20,21,41,53,55, as HD patients commonly have CAG repeat expansions that yield HTT
proteins with approximately forty residues in their polyQ domain2. In experimental studies of Q44-HTTex1 fibrils formed in vitro, similar to our analysis of the polyQ15 peptide fibrils
above, a large majority of the polyQ residues are observed to be buried in the fibril core, in a multi-nm-size block-like core architecture41. The polyQ-expanded HTTex1 protein fibrils were
observed to form protofilament architectures21,41 where the polyQ segment adopted a _β_-hairpin structure (Supplementary Fig. 1C)8. NMR on Q44-HTTex1 fibrils revealed a _β_-hairpin with a
single turn and two ~20-residue-long _β_-strands (Supplementary Fig. 1C). This experimental finding was enabled by isotopic dilution studies of Q44-HTTex1 done via NMR, showing close
intra-protein contacts between the “a”- and “b”-type strand backbones8. This finding recapitulated an earlier 2D-IR study that reported _β_-hairpins in longer polyQ peptide aggregates
lacking HTT flanking segments30. The presence of _β_-hairpins in (long) polyQ fibrils also was suggested by other mechanistic and mutational studies, in vitro and in cells56,57,58,59.
Solid-state NMR studies with site-specific amino acid labels have indicated that the ordered _β_-sheet core extends from the final N17-domain residue (F17) to the penultimate glutamine in
the polyQ segment20,28. Unlike the polyQ segment, the two polyQ-flanking segments of HTTex1 (see Fig. 1A) were found to be solvent exposed, lack _β_-structure, and display increased motion
and disorder19,21,22,23. The short N-terminal N17 segment has been reported to adopt a random coil or _α_-helical structure, depending on context28,60. In fibrils, electron paramagnetic
resonance (EPR) and ssNMR have shown the N17 segment to display partial order, and partly _α_-helical structure19,21,28. The longer C-terminal proline-rich domain (PRD) is disordered, except
for the polyproline-II (PPII) helices at the locations of the two oligoproline segments. Using these experimental data as input, we constructed the Q44-HTTex1 core base-architecture after
the schematic model described earlier8,20,21,41. The 44-residue polyQ segment has a _β_-hairpin conformation with a tight _β_-turn, containing an even number of residues, and two long
_β_-strands extending from F17 to the penultimate glutamine. A notable feature of polyQ protein aggregates is that the polyQ domains can come together in many different orientations and
alignments, yielding a large propensity for heterogeneity and stochatistic modes of assembly20,23,41. Here, a configuration was selected to construct a model that mimics our previous
schematic model41, with flanking domains evenly distributed on both sides of the fibril core. To this end, we attached the appropriate HTTex1 flanking domains to the terminal regions of the
polyQ section, thereby achieving the formation of monomer building blocks as illustrated in the top panel of Fig. 6A. The thus constructed HTTex1 fibril was subjected to unrestrained
all-atom MD simulations to assess its stability and monitor its structural dynamics. The fibril conformation obtained after a 5-_μ_s simulation is shown in Fig. 6A. STRUCTURAL ANALYSIS OF
HTTEX1 FIBRIL The resulting structure (Fig. 6A) reveals interesting features that permit comparison to experimental studies. Firstly, consistent with the polyQ15 fibril model, the HTTex1
polyQ core structure is found to be highly stable. A noteworthy observation lies in the _β_-turn conformation embedded within the _β_-hairpin structure: The simulation data underscores
predominance of the type II turn over a type I’ conformation (Supplementary Fig. 16), a finding that concurs with the experimental evidence gathered from ssNMR study of such compact turns8.
As for polyQ15 fibrils, a minority population of glutamines (the light-green side-chains of the middle image in Fig. 6A) is exposed to the solvent; these surface side-chains, as analyzed in
Figs. 4 and 5 above, show a semi-rigid behavior. The constrained dynamics of these solvent-exposed glutamines stand in large contrast to the dynamic disorder of both non-polyQ flanking
domains—whose disposition and structure are of substantial interest, given that they govern key structural and pathogenic properties of the protein and its aggregates19,20,21,41,61,62.
Figure 6A illustrates the high level of disorder that appears in the MD ensemble of the flanking domains, manifested in both the N17 and the PRD. Although such pronounced disorder interferes
with the detailed experimental study, its appearance in simulations fits with the dynamic disorder of this fuzzy coat observed in experiments, whether based on EPR, EM, or
ssNMR4,19,20,21,22,23. The secondary-structure preferences of N17, polyQ, and PRD are summarized in Supplementary Fig. 17. There has been significant interest especially in the N17 segment,
as it drives HTTex1 aggregation, but also harbors post-translational modifications that regulate HTTex1 (dis)aggregation and degradation63,64,65. The fate of N17 in the fibrils has remained
somewhat opaque, with seemingly conflicting reports of the presence and absence of (partial) _α_-helicity. The obtained HTTex1 fibril model provides relevant molecular insights, as its N17
segment displays a mixed secondary structure content, with much disorder (Fig. 6C and Supplementary Figs. 18–21). Virtually all N17 residues are seen to show some propensity for disorder,
such that a subset of the proteins in the modeled fibril has an N17 devoid of _α_-helical structure (Fig. 7D). Yet, in close to half of the protein monomers, an _α_-helix is observed within
N17. These findings match well to ssNMR analysis of the structure of N17 in fibrillar samples:21,28 Signals from an _α_-helical N17 were detected, but helicity was constrained to only part
of the segment (Fig. 6B). The helical residues seen experimentally coincide remarkably well with the residues found to favor helicity in our model (Fig. 6C), providing further support for
the validity of this structural ensemble. The observation that a significant part of N17 is not _α_-helical finds experimental support as well22,41,66. Thus, also this experimental finding
of N17 heterogeneity and plasticity is clearly recapitulated in the obtained fibril model: we observe an innate heterogeneity in structure and dynamics, even among protein monomers in the
same fibril (Fig. 7). C-terminal to the polyQ segment, the PRD is of biological interest given its role in reducing aggregation propensity, as well as its implication in toxic mechanisms. In
HTTex1 fibrils made in vitro, the PRD is known to display a gradient of dynamics19,20,21,22,41,66. Experimentally, the PRD is relatively rigid proximal to the polyQ core, whilst its very
tail end is highly flexible. The latter is evidenced by those residues showing up in INEPT-based MAS NMR measurements that are selective for highly flexible residues, such as the 2D
INEPT-TOBSY spectrum on Q44-HTTex1 fibrils in Fig. 6D. These data are consistent with prior studies using similar methods as well as relaxation measurements21,22,31. The MD simulations
indicate that the mobility of the PRD is not only constrained by its attachment to the rigid polyQ core, but also by pronounced PRD–PRD intermolecular interactions. Prior work has inferred a
propensity for such interactions, especially in context of filament–filament interactions21,32,41. The current model suggest that such interactions are also prominent in structuring the
flanking domains of isolated protofilaments. A notable feature of the HTTex1 fibril structural ensemble that was not a priori expected by these authors is that the flanking domains are not
showing much interaction with the polyQ amyloid core surface. Throughout the simulation, the flanking domains display substantial disorder, but preferentially cluster together. This leaves
the outer polyQ core surface easily accessible not only to solvent, but also to amyloid-binding molecules such as thioflavin-T (ThT) as well as PET ligands10. An in-depth and comprehensive
visualisation of the described HTTex1 fibril structure can be found in the Supplementary Movie 1. CAVEATS AND FIBRIL POLYMORPHISM It is important to note here that polyQ-based protein
aggregates display a persistent and characteristic structural heterogeneity that is impossible to fully capture in practical MD simulations. Prior studies have discussed the propensity for
polyQ segments to self-assemble in a disordered or stochastic fashion, due to the lack of sequence variation8,20,67. For instance, a polyQ chain extending an existing fibril can be added in
different orientations. This variability is to some extent already displayed in our model: The alternating monomers in the single sheet in Fig. 7D have differently organized _β_-hairpins in
their polyQ segment and their N17 segments on opposite sides of the filament. However, in real samples, one can expect a much more random patterning, which would vary at different locations
among even a single fibril. A similar stochastic feature that is explicitly missing from our model is that incoming polyQ segments can add to the fibril with register mis-alignments,
resulting in shorter or longer _β_-strands in slightly different sequence positions, without a major energetic cost. This would be expected to yield fibril cores with varying fibril widths,
local defects, and protein-to-protein structural variations20,23,67 not accessible to more canonical amyloid fibrils formed by other proteins. This includes the recently reported cryo-EM
structure of amyloid fibrils formed by the Orb2 protein (6VPS), a functional amyloid involved in memory formation (Supplementary Fig. 22)49. Although it is glutamine-rich in its amyloid
core, the observed structure matches the typical in-register parallel fold and its glutamine torsion angles differ from those of the two polyQ conformers (Supplementary Fig. 22B–C). The
presence of non-glutamine residues in the fibril core dictates a sequence-alignment that is absent in polyQ protein fibrils. In contrast, the latter fibrils are expected (and observed23,41)
to display inherent structural variations between fibrils in a single sample, and even within single fibrils. Thus it appears that, like snowflakes, each HTTex1 fibril is unique (which
prohibits the canonization of a single definitive protofilament structure) but still characterizable by well-defined structural features. This indistinguishability in local (atomic)
structure of differing fibril architectures manifests for instance in the near-identity of ssNMR peak positions reported for polymorphic HTTex1 fibrils. Indeed, like other amyloids, polyQ
proteins form different fibril polymorphs depending on experimental conditions21,32,68. This structural variation imbues the fibrils with different degrees of cytotoxicity32,68,69. Notably,
the HTTex1 polymorphs often reflect a type of ’supramolecular’ polymorphism, with different supramolecular assemblies formed from similarly structured protofilaments41. The current model is
expected to illuminate the atomic level conformation of one such protofilament, as it is based on experimental constraints that define its structure. Even inside a single protofilament, the
unusual block-like architecture of polyQ fibrils permits variations in the number of sheets packed into a single protofilament23. Here, we also fixed this parameter, based on the dominant
structures seen in one of our prior studies of Q44-HTTex1, but also this parameter certainly varies between and within samples. Thus, in summary, in numerous different ways, the structure of
HTTex1 is expected and observed to vary from sample to sample, from fibril to fibril, and even within single fibrils. This manifests in cryo-ET, cryo-EM, and AFM studies as fibrils that
show variability in their structure, with much less order than many other amyloid fibrils. Capturing this diversity in a single set of MD simulations is impractical, but we consider the
obtained structures as representative of the canonical or typical protofilaments seen in HTTex1 in vitro fibrils. IMPLICATIONS FOR HTT FIBRIL INTERACTIONS AND PROPERTIES A number of notable
features of our model match or rationalize reported biological properties of HTTex1 fibrils. We obtained important insights into the surface-accessible molecular features of the HTTex1
fibrils’ fuzzy coat. Fibril surface properties are crucial for their biological, possibly pathological, properties. In our fibril architecture, the N17 segment is found to reside outside the
fibril core, where it displays conformational and dynamic disorder. Crucially, however, N17 is tightly enclosed by the C-terminal PRD domains: Residues in N17 may be solvent-accessible, but
are nonetheless largely inaccessible to larger macromolecules such as chaperones, kinases, ubiquitinases, and other potential N17 interaction partners, as can be unambiguously concluded
from polymer brush theory (Fig. 8). Thus although in Fig. 7B the known phosphorylation sites T3, S13, and S16 may appear accessible, viewing this monomer in context (Figs. 7A, 8) underlines
that the N17 segment is fully surrounded by the longer PRD segments. Similarly, the only HTTex1 ubiquitination sites involve lysines K6, K9, and K15 in N17, rendering them largely
inaccessible in fibrillar HTTex1. The buried nature of N17 also rationalizes the low level of engagement by the TRiC chaperone:34,70 Although known to bind this part of HTTex1, prior EM
studies have shown TRiC to be unable to engage HTTex1 fibrils, except near the fibril ends. N17 is also implicated in membrane interactions, such that its preferential exposure at fibril
ends may explain the latter to be engaged with the ER membranes62,71,72. Let us briefly discuss the implications of our models for HTTex1 fibrils formed by proteins with longer (and shorter)
polyQ segments. Our HTTex1 fibril modeling focused on the HD-relevant Q44 that falls into the regime of common expansion lengths seen among patients. Yet, famously, mutant proteins can
differ widely in their polyQ lengths. As illustrated in the polyQ15 peptide fibril, and discussed elsewhere38,66,69, proteins with shorter polyQ lengths can still form fibrils (at least in
vitro). However, in such fibrils the polyQ segment may not form a _β_-hairpin, but instead occupy a single extended _β_-strand. Naturally, this would modulate the disposition of flanking
segments on the fibril surface. Nonetheless, the qualitative architecture would remain unchanged. Conversely, HTTex1 with longer polyQ lengths, such as those associated with juvenile HD,
would be expected to form amyloid cores featuring multiple turns, unlike the single-turn structures analyzed here for Q44-HTTex1. Naturally—although we expect the obtained fibril structure
to be a good representation of the fibrils present in our samples and also to have strong predictive and descriptive qualities for cellular HTTex1 aggregates—it cannot be excluded that
cellular factors (ranging from chaperones to membrane interactions) may modulate the aggregation mechanism to the extent that the mature fibril architecture differs from the one obtained in
vitro. Yet, unlike for other amyloid proteins, a remarkable feature of the HTTex1 ssNMR studies is that multiple groups have studied a variety of HTTex1 fibrils and always found the same
signature spectra that are connected to the structural parameters used to construct our model20,21,22,32,41. Indeed, there is little evidence for a qualitative change in fibril architecture.
Thus, we are inclined to expect that while cellular conditions may change certain details in the fibril structure, they would not fundamentally change the protofilament architecture. The
presented structural models of polyQ15 and Q44-HTTex1 amyloid fibrils provide the best atomistic views of these disease-associated protein inclusions to date, derived from a multi-technique
structural analysis through integrative modeling. The HTTex1 structure rationalizes a variety of experimental findings with notable biological and biomedical implications. The polyQ segment
is mostly buried within the fibril, but the model reveals the structural and dynamical features of the minority of solvent-facing residues. These surface residues have proved challenging for
experimental study, necessitating tailored and targeted approaches in, e.g., ssNMR53. A better structural understanding of this special polyQ surface will be useful in efforts to design
polyQ-amyloid-specific binders, e.g., for PET imaging10. The visualization of the dynamically and structurally heterogeneous flanking domains enhance our understanding of their accessibility
in the fibrils and pave the way for more in-depth analyses of their role in intracellular interactions with proteins and organelles. METHODS PROTEIN PRODUCTION AND FIBRILLATION Mutant
huntingtin exon 1 with a 44-residue polyQ core was expressed as part of a maltose binding protein (MBP) fusion protein, with the MBP attached to the N-terminus of HTTex1 to prevent
aggregation20,21. The fusion protein MBP-Q44-HTTex1 was expressed in _Escherichia coli_ BL21 (DE3) pLysS cells (Invitrogen, Grand Island, NY). Uniformly 13C and 15N labeled MBP-Q44-HTTex1
protein was expressed with 13C D-glucose and 15N ammonium chloride for MAS ssNMR studies. Then, cells were pelleted at 7 000 g, resuspended in phosphate buffered saline (PBS), pH 7.4 and
lysed in presence of 1 mM phenylmethanesulfonyl fluoride (PMSF) by a HPL 6 (Maximator Benelux BV, The Netherlands). After that, cells were centrifuged at 125,000 × _g_ for 1 h using an
Optima LE-80K ultra-centrifuge (Beckmann Coulter). The supernatant was filtered over Millex-GP syringe-driven 0.22 μm PES membranes (Millipore Sigma, Burlington, MD). The MBP-Q44-HTTex1
protein was purified by fast protein liquid chromatography (FPLC) using a 5 ml HisTrap HP nickel column (GE Healthcare, Uppsala, Sweden) with 0.5 M imidazole gradient (SKU I5513-100G, Sigma,
St. Louis, MO) on an AKTA system (GE Healthcare, Chicago, IL). The imidazole was removed from the purified protein using an Amicon Ultra centrifugal filter with a regenerated cellulose
membrane (Millipore Sigma, Burlington, MA). At least 3 washes with imidazole-free PBS buffer were done. Protein concentration was calculated from the absorbance at 280 nm. According to
ProtParam tool by ExPasy73 the extinction coefficient of the fusion protein is 66,350 M−1cm−1. Protein aggregation was initiated by addition of Factor Xa protease (SKU PR-V5581, Promega,
Madison, WI) at 22 °C, in order to cleave off the MBP fusion tag21,41 and release Q44-HTTex1. To prepare the ΔN15-Q44-HTTex1 samples, trypsin protease was used to sever the fusion protein41,
yielding fibrils in which most of the N17 segment is absent. After 3 days, the obtained mature fibrils were washed with PBS to remove the cleaved MBP. SAMPLES FOR DEUTERIUM EXCHANGE MAS NMR
STUDIES For the proton-deuterium-exchange (HDX) experiments, prior to cleavage, batches of the fusion protein were exchanged into either protonated or deuterated PBS buffer by solvent
exchange (using Amicon centrifugal filters with 10 kDa cutoff). Next, the protein concentration was adjusted to 50 μM (with protonated or deuterated PBS, respectively). Factor Xa was added
(1:400 molar ratio, protease:fusion protein), to permit three days of aggregation at 37 °C. Next, the protonated or deuterated fibrils were recovered and washed with matching PBS buffer.
Just before the MAS NMR measurements, the fibrils were washed with PBS, either protonated or deuterated, to study the proton-deuterium-exchange process by MAS NMR. POLYQ PEPTIDE SAMPLES For
EM and NMR experiments, synthetic polyQ-based peptides were prepared and studied in their aggregated state. These peptides were obtained by solid-phase peptide synthesis (SPPS) from
commercial sources, submitted to disaggregation protocols and permitted to aggregate in PBS buffer8. The aggregated polyQ15 peptide51 studied by TEM had the sequence D2Q15K2, synthesized by
SPPS by WatsonBio (Houston, TX). The polyQ peptide used in the 15N-detected 2D NMR studies (Supplementary Fig. 7) had the sequence K2Q11pGQ11K2 (p indicates D-proline), with two sequential
Gln residues outfitted with uniform 13C and 15N labeling, synthesized by the Yale University peptide facility8. The peptides were aggregated at 1 mg/mL concentration in PBS buffer (pH 7.4)
at 37∘C. Aggregates were harvested by centrifugation after 2–3 weeks. TRANSMISSION ELECTRON MICROSCOPY (TEM) Transmission electron microscopy (TEM) was performed on mature D2Q15K2 peptide
fibrils and Q44-HTTex1 fibrils. The fibrils were re-suspended in MiliQ and then 5 _μ_l of the sample was deposited on the plain carbon support film on 200 mesh copper grid (SKU FCF200-Cu-50,
Electron Microscopy Sciences, Hatfield, PA). The grid was glow discharged for 0.5–1 min before adding the sample. After 30 s of the sample deposition, the excess MiliQ was removed by
blotting, and immediately the negative staining agent 1% (w/v) uranyl acetate was applied. After 0.5–1 min, the excess stain was removed and the grid was air dried. The images were recorded
on a Tecnai T12 or CM12 transmission electron microscope. NMR EXPERIMENTS The hydrated U-13C,15N-labeled Q44-HTTex1 fibrils were packed into a 3.2 mm ssNMR rotor (Bruker Biospin) using a
ultracentrifugal packing tool74. The fibrils were packed in the rotor by centrifugation at ≈130,000 × _g_ in a Beckman Coulter Optima LE-80K ultracentrifuge equipped with an SW-32 Ti rotor
for 1 h. Experiments were performed on a wide-bore Bruker Avance-I 600 MHz (14.1 T) spectrometer or Bruker Avance Neo 600 MHz (14.1 T) spectrometer, using triple-channel (HCN) 3.2 mm MAS
EFree probes from Bruker. Data acquisition was done using Bruker Topspin software (version 4.1.3). Chemical shifts were indirectly referenced based on the 13C shifts of adamantane, and
reported relative to aqueous DSS (13C, 1H) or liquid ammonia (15N). NMR spectra were processed, analyzed and plotted using NMRpipe (version 2019.217.13.13) and CcpNmr Analysis software
(version 2.4)75. All experiments were acquired using two-pulse phase modulated (TPPM) proton decoupling of 83 kHz during acquisition76. The 2D 13C–13C DARR experiments77 on uniformly labeled
Q44-HTTex1 fibrils (Fig. 1F) were performed using a 3-μs 90∘ pulse on 1H, 4-_μ_s 90∘ pulses on 13C, a 1H−13C CP contact time of 1 ms at 275 K, a DARR mixing time of 25 ms, and a recycle
delay of 2.8 s. 2D NCA and NCO experiments were performed on U-13C,15N-labeled Q44-HTTex1 fibrils, as follows. 2D 13C–15N NCO experiments (Fig. 1G) were done at 277 K using a 3-_μ_s 90∘
pulse on 1H, 8-_μ_s 180∘ pulse on 13C, 1H–15N contact time of 1.5 ms, 15N–13C contact time of 4 ms and recycle delay of 2.8 s. 2D 13C–15N NCA experiments were done at 277 K using a recycle
delay of 2.8 s, a 3-_μ_s 90∘ pulse on 1H, 8-_μ_s 180∘ pulse on 13C, 1.5 ms and 4 ms 1H–15N and 15N–13C contact times, respectively. In NCA and NCO experiments, the power levels for 15N and
13C during N–C transfer steps were 50 kHz and 62.5 kHz, respectively. The 2D refocused-INEPT 13C–13C 2D spectrum of U-13C,15N-labeled Q44-HTTex1 fibrils (Fig. 6D) was obtained with total
through-bond correlation spectroscopy (TOBSY; P931) recoupling, measured at MAS rate of 8.3 kHz, using a 6 ms of TOBSY mixing time, a 3-μs 90∘ pulse on 1H, 4-μs 90∘ on 13C, at a temperature
of 275 K78. To observe and assign the backbone and side-chain protons of the fibril polyQ core, 1H–15N heteronuclear correlation (HETCOR) experiments were performed on both polyQ model
peptides (where only Gln were labeled) and fully 13C,15N-labeled HTTex1 fibril samples. The former sample allows us to exclude contributions from non-Gln residues to the detected signals.
These experiments were applied to aggregates of K2Q11pGQ11K2 peptides in which only two (sequential) Gln were labeled with 13C,15N, which were previously shown to display the characteristic
polyQ core signature8. To compare those to the polyQ core of HTTex1 fibrils, we employed aggregates formed from U13C,15N Q44-HTTex1 cleaved with trypsin (Supplementary Fig. 7). The 1H–15N
HETCOR experiments on both samples were done using a MAS rate of 13 kHz, 100 and 350 _μ_s CP contact times, 4 s recycle delay, a 3-_μ_s 90∘ 1H pulse, and at 275 K temperature. 100 kHz
homonuclear FSLG 1H decoupling was applied during the t1 evolution time. For the peptide fibrils, 128 scans (per t1 point) were acquired; for the protein fibrils 64 scans. The HDX MAS NMR
experiments were performed using a 700 MHz Bruker NMR spectrometer, equipped with a 1.3 mm fast-MAS HCN probe. 2D 15N–1H spectra were obtained using 2 ms 1H–15N and 1 ms 15N–1H CP transfers
and a recycle delay of 1.1 s. Relaxation measurements were performed at 60 kHz MAS using relaxation delays of 0, 10, 30, 50, 100, and 200 ms for 1H–15N _T_1_ρ_ measurements using a 1H–15N
spin lock amplitude of 18 kHz, and relaxation delays of 0, 0.1, 1, 2, 4, 8, and 16 s for 1H–15N _T_1 measurements. The _T_1/_T_1_ρ_ trajectories were fit to single exponentials. MD
SIMULATIONS Let us first describe general simulation details. All MD simulations were carried out on the fast, free, and flexible Gromacs engine79. All systems were first energy-minimized
using steepest descent with one conjugate gradient step every 100 steps, then equilibrated through a 200-ps MD simulation in the NVT ensemble with positional restraints (1 000 kJ/mol/nm2) on
heavy atoms, followed by three 100-ps NPT runs with positional restraints (1000, 500, and 100 kJ/mol/nm2) on heavy atoms. (To create independent replicates of the 30 polyQ amyloid core
lattices, see Supplementary Fig. 4, we alternatively equilibrated them using dihedral restraints on the _χ_1 and _χ_3 angles: First through a 100-ps NVT run with 1 000 kJ/mol/rad2, followed
by four 100-ps NPT runs with 1 000, 500, 250, and 100 kJ/mol/rad2.) The production MD simulations were done in the NPT ensemble, obtained through the Bussi–Donadio–Parrinello80 thermostat (T
= 300 K, _τ_T = 0.2 ps) and the isotropic (for the polyQ lattice systems) or semi-isotropic (Q15 and HTTex1 systems, _x__y_ and _z_ coupled separately) Parrinello–Rahman81 (P = 1 bar, _τ_P
= 2 ps, _κ_P = 4.5 × 10−5 bar−1) barostat. The van der Waals interactions were switched off between 1.0 and 1.2 nm; long-range electrostatics were treated via Particle Mesh Ewald82,83 with
fourth-order interpolation, a real-space cut-off at 1.2 nm, and size-optimized fast Fourier transform parameters (grid spacing of roughly 0.16 nm). Covalent bonds involving hydrogens were
constrained to their equilibrium lengths by (fourth-order double-iteration) parallel linear constraint solver (P-LINCS)84. Timestep was 2 fs, Verlet neighbor lists updated every 20 fs with
the neighbor list radius automatically determined. Input files containing the complete sets of simulation parameters used are permanently openly available on Zenodo85. Let us then describe
the specific details for polyQ amyloid core lattices. We first describe building these systems. To build atomistic-resolution models of the internal structure of the polyQ amyloid core, we
assumed it to comprise a lattice of antiparallel _β_-sheets stacked such that the Gln side-chains interdigitate. The pairs of side-chain dihedral angles (_χ_1, _χ_3) were set to satisfy the
known characteristic of the interdigitating polyQ amyloid core:8,26 the existence of side-chain–side-chain hydrogen bond interactions. We took the fibril-axis direction to align with the
Cartesian coordinate _z_, and to be perpendicular to the _β_-strands (aligned with _x_). To construct the 3D lattice, we considered a minimal unit cell consisting of eight Gln residues
arranged in a 2 × 2 × 2 pattern (Fig. 2A): Along _x_, the unit cell contains a minimal peptide chain segment of two amino acids (2 × 2 × 2), representing the alternating (odd/even, i.e.,
pointing above and below the _β_-sheet plane) residues of the _β_-strand. Along _z_, to describe an antiparallel _β_-sheet, minimum two _β_-strands are needed (2 × 2 × 2); in contrast to
parallel in-register sheet structures that could be represented with a single repeating _β_-strand. Along _y_, the unit cell contains two distinct neighboring _β_-sheets to permit and probe
distinct sheet–sheet interfaces (2 × 2 × 2). Figure 2A illustrates how each eight-Gln unit cell contains four Gln–Gln pairs, which establish backbone hydrogen bonds (shown in purple) as well
as side-chain hydrogen bonds (in orange). All the hydrogen bonds are aligned roughly along the fibril axis, _z_. For each Gln–Gln pair, there are 8 distinct classes of (_χ_1, _χ_3)
orientations that permit side-chain–side-chain hydrogen bond chains along _z_ (see Supplementary Fig. 2). As there are 4 Gln–Gln pairs in the unit cell, there are 84 = 4 096 possible
plausible atomistic structures of the 8-Gln unit cell. Accounting for rotational and translational symmetry of the 3D lattice reveals, however, that at most 1 280 of these structures are
unique. An important further consideration is that ssNMR experiments conclusively demonstrate that consecutive residues within each _β_-strand must adopt the same backbone conformations, as
indicated by identical chemical shifts. As a result, each distinct strand exclusively contains either type “a” or type “b” Gln residues (Fig. 1H). After applying this filter, the number of
possible distinct unit cells is reduced to 30. For each of these 30 unit cells candidates, we performed all-atom MD simulations. The fully periodic MD simulation box was filled by 40
identical unit cells, that is, a total of 8 × 40 = 320 Gln residues, organized as a stack (along _y_) of 4 antiparallel _β_-sheets, with each sheet comprising 8 _β_-strands of periodic
(along _x_) Q10 peptides, see also Supplementary Table 1. Let us then describe the MD simulation details specific to polyQ amyloid core lattice simulations. Acknowledging the limitations of
a classical mechanics approximation (force field) of an inherently quantum system, we employed in parallel three state-of-the-art MD force fields (one from each of the main force field
families): AMBER14SB86, OPLSAA/M87, and CHARMM36m88. Gromacs version 2018.3 was used. During the production runs (10 _μ_s for position-restraint-initialized Amber14SB (Fig. 2B), 1 _μ_s for
other force fields and/or dihedral-restraint initialization (Supplementary Fig. 4)), the stability of the 30 structural candidates was evaluated at 5 ns, 100 ns, 200 ns, and 1 _μ_s. At each
of these time points, only the candidates that maintained their stability above 0.9 (see Eq. (1)) were further continued. The stability _S_(_t_) of the given structural candidate at
simulation time _t_ was defined based on the _χ_1(_t_) and _χ_3(_t_) dihedral angles compared to the initial energy-minimized MD structure: $$S(t)=\frac{{N}_{1}(t)+{N}_{3}(t)}{2N},$$ (1)
where _N_ = 320 is the total number of residues, and _N__i_(_t_), _i_ = {1, 3} is the number of residues whose _χ__i_(_t_) is within 90∘ of its initial value at time _t_. For the stable
candidates _M1_ and _M2_, the complete production trajectories (with frames saved every 2 ps) were used for analysis. Let us then describe the MD simulation details of the solvated polyQ15
fibril systems. Q15 peptide fibrils were constructed from each of the two stable ssNMR-verified core models, _M1_ and _M2_. Two successive aspartic acid (DD) residues were added to the
N-terminus and two lysines (KK) to the C-terminus of the Q15 peptide to mimic the D2Q15K2 peptide widely studied by experiments8,24,27,50. The N-terminus was further capped with an acetyl
(Ace) group, and the C-terminus was set uncharged (with –COOH capping) to match the peptides used in our experiments. A 7-sheet fibril structure was constructed, with each sheet comprising
eight Ace-D2Q15K2 peptides. The simulation box was set up to form a quasi-infinite fibril (along _z_) under periodic boundary conditions and solvated with ~9 800 water molecules in a cuboid
box of ~11 × 11 × 3.8 nm3, see also Supplementary Table 1. State-of-the-art MD force fields AMBER14SB86, OPLSAA/M87, and the TIP3P89 water model were used for calculations carried out on
the Gromacs version 2021.3. Production run length was 1 _μ_s; the complete production trajectories (with frames saved every 2 ps) were used for analysis. Let us then describe the MD
simulation details of the solvated Q44-HTTex1 fibril. Let us start with creating the system. The ssNMR data suggest that the HTTex1 aggregates display the same spectral patterns observed in
polypeptide polyQ fibrils8,38. This indicates the presence of a common atomic polyQ core structure among them. Consequently, we constructed an atomistic-resolution structure of the mutant
HTTex1 fibril utilizing the polyQ core model described in the preceding section. In contrast to fibrils composed of short polyQ segments, the Q44-HTTex1 fibrils exhibited a distinct
structural characteristic, consisting of a _β_-hairpin structure with a single turn. Hence, the constructed core domain of our Q44-HTTex1 encompassed a stack of seven antiparallel
_β_-sheets, each sheet comprising twenty 44-residue polyQ hairpins (Q44). The hairpin arms consisted of “a" and “b" _β_-strands connected by a two-residue _β_-turn modeled based on
a type-I’ tight turn, known for its strong preference towards adopting a _β_-hairpin structure (1KH0)90. The N-terminus of HTTex1, comprising 17 residues, was modeled utilizing the crystal
structure of a single HTT(1–17) peptide in complex with the C4 single-chain Fv antibody (4RAV)60. The residue F17 was included within the _β_-sheet core, pairing with the penultimate
glutamine residue, as supported by the findings of ssNMR investigations28. The PRD domain of the HTTex1, including 50 residues (P62–P111), was modeled as an end-to-end-distance-maximized
random coil, with the two oligoproline (P62–P72 and P90–P99) segments in a polyproline-II-helix conformation. The terminal cappings were set to \({{\rm{NH}}}_{3}^{+}\) and COO− for the N and
C termini, respectively. The turns of neighboring Q44 hairpins were positioned on opposite sides of the fibril (Fig. 6A). The system was solvated with ~840 422 water molecules, resulting
in a total of ~2,800,000 atoms, in a cuboid box of ~38 × 38 × 19 nm3, see also Supplementary Table 1. The production MD simulations were then conducted for a duration of 5 μs (with frames
saved every 20 ps) on Gromacs version 2021.4 with AMBER14SB86 protein force field and TIP3P89 water. The calculation of dihedral angles, distances, and protein secondary structures were done
using the gmx_angle, gmx_distance (with gmx_traj used to obtain centers of mass for distances between groups of atoms), and gmx_do_dssp tools of GROMACS versions 2018.3 and 2021.4. Images
of molecular structures were created using VMD 1.9.3;91 except for Fig. 7 and the Supplementary Movie 1, which were created using ChimeraX 1.792. POLYMER BRUSH THEORY The largest particle
that a polymer brush in an ideal solvent can accommodate has the size93,94 $${b}_{\max }=\root{4}\of {\frac{N{a}^{2}}{2{\pi }^{3}\sigma }},$$ (2) where _N_ is the number of monomer units per
polymer, _a_ the monomer size, and _σ_ the grafting density. For a Q44-HTTex1 amyloid fibril, _N_ ≈ 50 and _a_ ≈ 0.4 nm are the number of residues in the PRD domain and the persistence
length of an intrinsically disordered protein, respectively;95 and _σ_ ≈ 0.7 nm−2 is the density of PRD domains on the fibril sides where the polyQ-flanking segments reside. Using these
values results in \({b}_{\max }\approx 0.7\,{\rm{nm}}\); allowing for a good95 instead of an ideal solvent gives the \({b}_{\max }\approx 0.8\,{\rm{nm}}\) used in Fig. 8. REPORTING SUMMARY
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Experimental and simulation data generated in this
study have been deposited on the associated Zenodo repository85 13926360 that also contains the simulation input files and the initial and final coordinate files, as well as the Source Data
files for Figs. 1F; 2B,D,G; 3C; 4B,C; 5; and 6B–D. Relevant chemical shifts are in the BMRB 27045 and BMRB 25146. In this work the following existing protein structures were used: 4RAV,
1C4Z, 1UBQ, 6CQ0, 6VPS, and 1KH0. CODE AVAILABILITY The associated permanently openly available Zenodo repository85 13926360 contains the in-house Python scripts used for MD analysis.
REFERENCES * Ross, C. A. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. _Neuron_ 35, 819–822 (2002). Article CAS PubMed MATH
Google Scholar * Bates, G. P. et al. Huntington disease. _Nat. Rev. Dis. Prim._ 1, 1–21 (2015). Google Scholar * DiFiglia, M. et al. Aggregation of huntingtin in neuronal intranuclear
inclusions and dystrophic neurites in brain. _Science_ 277, 1990–1993 (1997). Article CAS PubMed MATH Google Scholar * Bäuerlein, F. J. et al. In situ architecture and cellular
interactions of polyQ inclusions. _Cell_ 171, 179–187 (2017). Article PubMed MATH Google Scholar * Peskett, T. R. et al. A liquid to solid phase transition underlying pathological
huntingtin exon1 aggregation. _Mol. Cell_ 70, 588–601.e6 (2018). Article PubMed PubMed Central Google Scholar * Galaz-Montoya, J. G., Shahmoradian, S. H., Shen, K., Frydman, J. &
Chiu, W. Cryo-electron tomography provides topological insights into mutant huntingtin exon 1 and polyQ aggregates. _Commun. Biol._ 4, 849 (2021). Article CAS PubMed PubMed Central
Google Scholar * Sawaya, M. R., Hughes, M. P., Rodriguez, J. A., Riek, R. & Eisenberg, D. S. The expanding amyloid family: Structure, stability, function, and pathogenesis. _Cell_ 184,
4857–4873 (2021). Article CAS PubMed PubMed Central Google Scholar * Hoop, C. L. et al. Huntingtin exon 1 fibrils feature an interdigitated _β_-hairpin–based polyglutamine core. _Proc.
Natl. Acad. Sci._ 113, 1546–1551 (2016). Article ADS CAS PubMed PubMed Central MATH Google Scholar * Scherzinger, E. et al. Huntingtin-encoded polyglutamine expansions form
amyloid-like protein aggregates in vitro and in vivo. _Cell_ 90, 549–558 (1997). Article CAS PubMed MATH Google Scholar * Liu, L. et al. Imaging mutant huntingtin aggregates:
Development of a potential PET ligand. _J. Med. Chem._ 63, 8608–8633 (2020). Article CAS PubMed MATH Google Scholar * van der Wel, P. C. A. Insights into protein misfolding and
aggregation enabled by solid-state NMR spectroscopy. _Solid State Nucl. Magn. Reson._ 88, 1–14 (2017). Article ADS PubMed PubMed Central Google Scholar * Fitzpatrick, A. W. &
Saibil, H. R. Cryo-EM of amyloid fibrils and cellular aggregates. _Curr. Opin. Struct. Biol._ 58, 34–42 (2019). Article CAS PubMed PubMed Central MATH Google Scholar * Chen, S.,
Ferrone, F. A. & Wetzel, R. Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. _Proc. Natl. Acad. Sci._ 99, 11884–11889 (2002). Article ADS CAS PubMed
PubMed Central Google Scholar * Guo, Q. et al. The cryo-electron microscopy structure of huntingtin. _Nature_ 555, 117–120 (2018). Article ADS CAS PubMed PubMed Central MATH Google
Scholar * Harding, R. J. et al. Huntingtin structure is orchestrated by HAP40 and shows a polyglutamine expansion-specific interaction with exon 1. _Commun. Biol._ 4, 1374 (2021). Article
CAS PubMed PubMed Central MATH Google Scholar * Lunkes, A. et al. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear
inclusions. _Mol. Cell_ 10, 259–269 (2002). Article CAS PubMed Google Scholar * Sathasivam, K. et al. Aberrant splicing of _HTT_ generates the pathogenic exon 1 protein in Huntington
disease. _Proc. Natl. Acad. Sci._ 110, 2366–2370 (2013). Article ADS CAS PubMed PubMed Central MATH Google Scholar * Neueder, A. et al. The pathogenic exon 1 HTT protein is produced
by incomplete splicing in Huntington’s disease patients. _Sci. Rep._ 7, 1307 (2017). Article ADS PubMed PubMed Central MATH Google Scholar * Bugg, C. W., Isas, J. M., Fischer, T.,
Patterson, P. H. & Langen, R. Structural features and domain organization of huntingtin fibrils. _J. Biol. Chem._ 287, 31739–31746 (2012). Article CAS PubMed PubMed Central Google
Scholar * Hoop, C. L. et al. Polyglutamine amyloid core boundaries and flanking domain dynamics in huntingtin fragment fibrils determined by solid-state nuclear magnetic resonance.
_Biochemistry_ 53, 6653–6666 (2014). Article CAS PubMed MATH Google Scholar * Lin, H.-K. et al. Fibril polymorphism affects immobilized non-amyloid flanking domains of huntingtin exon1
rather than its polyglutamine core. _Nat. Commun._ 8, 1–12 (2017). Article ADS Google Scholar * Isas, J. M., Langen, R. & Siemer, A. B. Solid-state nuclear magnetic resonance on the
static and dynamic domains of huntingtin exon-1 fibrils. _Biochemistry_ 54, 3942–3949 (2015). Article CAS PubMed Google Scholar * Nazarov, S., Chiki, A., Boudeffa, D. & Lashuel, H.
A. Structural basis of huntingtin fibril polymorphism revealed by cryogenic electron microscopy of exon 1 HTT fibrils. _J. Am. Chem. Soc._ 144, 10723–10735 (2022). Article CAS PubMed
Google Scholar * Perutz, M., Staden, R., Moens, L. & De Baere, I. Polar zippers. _Curr. Biol._ 3, 249–253 (1993). Article CAS PubMed Google Scholar * Perutz, M. F., Finch, J. T.,
Berriman, J. & Lesk, A. Amyloid fibers are water-filled nanotubes. _Proc. Natl. Acad. Sci._ 99, 5591–5595 (2002). Article ADS CAS PubMed PubMed Central Google Scholar * Sikorski,
P. & Atkins, E. New model for crystalline polyglutamine assemblies and their connection with amyloid fibrils. _Biomacromolecules_ 6, 425–432 (2005). Article CAS PubMed MATH Google
Scholar * Sharma, D., Shinchuk, L. M., Inouye, H., Wetzel, R. & Kirschner, D. A. Polyglutamine homopolymers having 8–45 residues form slablike _β_-crystallite assemblies. _Proteins:
Struct., Funct., Bioinf._ 61, 398–411 (2005). Article CAS Google Scholar * Sivanandam, V. et al. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. _J. Am.
Chem. Soc._ 133, 4558–4566 (2011). Article CAS PubMed PubMed Central MATH Google Scholar * Schneider, R. et al. Structural characterization of polyglutamine fibrils by solid-state NMR
spectroscopy. _J. Mol. Biol._ 412, 121–136 (2011). Article CAS PubMed MATH Google Scholar * Buchanan, L. E. et al. Structural motif of polyglutamine amyloid fibrils discerned with
mixed-isotope infrared spectroscopy. _Proc. Natl. Acad. Sci._ 111, 5796–5801 (2014). Article ADS CAS PubMed PubMed Central MATH Google Scholar * Caulkins, B. G., Cervantes, S. A.,
Isas, J. M. & Siemer, A. B. Dynamics of the Proline-Rich C-Terminus of Huntingtin Exon-1 Fibrils. _J. Phys. Chem. B_ 122, 9507–9515 (2018). Article CAS PubMed PubMed Central Google
Scholar * Mario Isas, J. et al. Huntingtin fibrils with different toxicity, structure, and seeding potential can be interconverted. _Nat. Commun._ 12, 4272 (2021). Article ADS CAS PubMed
PubMed Central MATH Google Scholar * Xiong, K., Punihaole, D. & Asher, S. A. UV resonance Raman spectroscopy monitors polyglutamine backbone and side chain hydrogen bonding and
fibrillization. _Biochemistry_ 51, 5822–5830 (2012). Article CAS PubMed Google Scholar * Shahmoradian, S. H. et al. TRiC’s tricks inhibit huntingtin aggregation. _eLife_ 2, e00710
(2013). Article PubMed PubMed Central Google Scholar * Wagner, A. S. et al. Self-assembly of mutant huntingtin exon-1 fragments into large complex fibrillar structures involves nucleated
branching. _J. Mol. Biol._ 430, 1725–1744 (2018). Article CAS PubMed MATH Google Scholar * van der Wel, P. C. A. Solid-state nuclear magnetic resonance in the structural study of
polyglutamine aggregation. _Biochem. Soc. Trans._ 52, 719–731 (2024). Article PubMed PubMed Central MATH Google Scholar * Rout, M. P. & Sali, A. Principles for integrative
structural biology studies. _Cell_ 177, 1384–1403 (2019). Article CAS PubMed PubMed Central MATH Google Scholar * Matlahov, I. & van der Wel, P. C. A. Conformational studies of
pathogenic expanded polyglutamine protein deposits from Huntington’s disease. _Exp. Biol. Med._ 244, 1584–1595 (2019). Article CAS Google Scholar * Sharma, D., Sharma, S., Pasha, S. &
Brahmachari, S. K. Peptide models for inherited neurodegenerative disorders: conformation and aggregation properties of long polyglutamine peptides with and without interruptions. _FEBS
Lett._ 456, 181–185 (1999). Article CAS PubMed MATH Google Scholar * Margittai, M. & Langen, R. Fibrils with parallel in-register structure constitute a major class of amyloid
fibrils: molecular insights from electron paramagnetic resonance spectroscopy. _Q. Rev. Biophys._ 41, 265–297 (2008). Article CAS PubMed Google Scholar * Boatz, J. C. et al.
Protofilament structure and supramolecular polymorphism of aggregated mutant huntingtin exon 1. _J. Mol. Biol._ 432, 4722–4744 (2020). Article CAS PubMed PubMed Central MATH Google
Scholar * Punihaole, D., Workman, R. J., Hong, Z., Madura, J. D. & Asher, S. A. Polyglutamine Fibrils: New Insights into Antiparallel _β_-Sheet Conformational Preference and Side Chain
Structure. _J. Phys. Chem. B_ 120, 3012–3026 (2016). Article CAS PubMed Google Scholar * van der Wel, P. C. A. Dihedral angle measurements for structure determination by biomolecular
solid-state NMR spectroscopy. _Front. Mol. Biosci._ 8, 791090 (2021). Article PubMed PubMed Central Google Scholar * Lovell, S. C., Word, J. M., Richardson, J. S. & Richardson, D. C.
The penultimate rotamer library. _Proteins_ 40, 389–408 (2000). Article CAS PubMed MATH Google Scholar * Chan, J. C. C., Oyler, N. A., Yau, W.-M. & Tycko, R. Parallel β-sheets and
polar zippers in amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p. _Biochemistry_ 44, 10669–10680 (2005). Article CAS PubMed Google Scholar * Wiegand, T. et al.
Asparagine and Glutamine Side-Chains and Ladders in HET-s(218–289) Amyloid Fibrils Studied by Fast Magic-Angle Spinning NMR. _Front. Mol. Biosci._ 7, 582033 (2020). Article CAS PubMed
PubMed Central Google Scholar * Wagner, G., Pardi, A. & Wuethrich, K. Hydrogen bond length and 1H NMR chemical shifts in proteins. _J. Am. Chem. Soc._ 105, 5948–5949 (1983). Article
CAS Google Scholar * Hori, S., Yamauchi, K., Kuroki, S. & Ando, I. Proton NMR chemical shift behavior of hydrogen-bonded amide proton of glycine-containing peptides and polypeptides as
studied by ab initio MO calculation. _Int. J. Mol. Sci._ 3, 907–913 (2002). Article CAS Google Scholar * Hervas, R. et al. Cryo-EM structure of a neuronal functional amyloid implicated
in memory persistence in _Drosophila_. _Science_ 367, 1230–1234 (2020). Article ADS CAS PubMed PubMed Central MATH Google Scholar * Perutz, M. F., Johnson, T., Suzuki, M. & Finch,
J. T. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. _Proc. Natl. Acad. Sci._ 91, 5355–5358 (1994). Article ADS CAS PubMed PubMed
Central Google Scholar * Smith, A. N. et al. Structural Fingerprinting of Protein Aggregates by Dynamic Nuclear Polarization-Enhanced Solid-State NMR at Natural Isotopic Abundance. _J. Am.
Chem. Soc._ 140, 14576–14580 (2018). Article CAS PubMed PubMed Central MATH Google Scholar * Perutz, M. F., Pope, B. J., Owen, D., Wanker, E. E. & Scherzinger, E. Aggregation of
proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid β-peptide of amyloid plaques. _Proc. Natl. Acad. Sci. USA_
99, 5596–5600 (2002). Article ADS CAS PubMed PubMed Central Google Scholar * Matlahov, I., Boatz, J. C. & van der Wel, P. C. Selective observation of semi-rigid non-core residues
in dynamically complex mutant huntingtin protein fibrils. _J. Struct. Biol.: X_ 6, 100077 (2022). PubMed Google Scholar * Jayaraman, M. et al. Slow amyloid nucleation via _α_-helix-rich
oligomeric intermediates in short polyglutamine-containing huntingtin fragments. _J. Mol. Biol._ 415, 881–899 (2012). Article CAS PubMed MATH Google Scholar * Poirier, M. A. et al.
Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. _J. Biol. Chem._ 277, 41032–41037 (2002). Article CAS PubMed MATH Google Scholar * Thakur, A. K.
& Wetzel, R. Mutational analysis of the structural organization of polyglutamine aggregates. _Proc. Natl. Acad. Sci._ 99, 17014–17019 (2002). Article ADS CAS PubMed PubMed Central
Google Scholar * Zhang, Q. C. et al. A compact β model of huntingtin toxicity. _J. Biol. Chem._ 286, 8188–8196 (2011). Article CAS PubMed PubMed Central MATH Google Scholar * Kar, K.
et al. β-hairpin-mediated nucleation of polyglutamine amyloid formation. _J. Mol. Biol._ 425, 1183–1197 (2013). Article CAS PubMed PubMed Central MATH Google Scholar * Kandola, T. et
al. Pathologic polyglutamine aggregation begins with a self-poisoning polymer crystal. _eLife_ 12, RP86939 (2023). Article CAS PubMed PubMed Central Google Scholar * De Genst, E. et al.
Structure of a single-chain Fv bound to the 17 N-terminal residues of huntingtin provides insights into pathogenic amyloid formation and suppression. _J. Mol. Biol._ 427, 2166–2178 (2015).
Article PubMed PubMed Central MATH Google Scholar * Duennwald, M. L., Jagadish, S., Muchowski, P. J. & Lindquist, S. L. Flanking sequences profoundly alter polyglutamine toxicity in
yeast. _Proc. Natl. Acad. Sci._ 103, 11045–11050 (2006). Article ADS CAS PubMed PubMed Central Google Scholar * Burke, K. A., Kauffman, K. J., Umbaugh, C. S., Frey, S. L. &
Legleiter, J. The interaction of polyglutamine peptides with lipid membranes is regulated by flanking sequences associated with huntingtin. _J. Biol. Chem._ 288, 14993–15005 (2013). Article
CAS PubMed PubMed Central Google Scholar * Thakur, A. K. et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. _Nat. Struct.
Mol. Biol._ 16, 380–389 (2009). Article CAS PubMed PubMed Central Google Scholar * Gu, X. et al. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin
induced disease pathogenesis in HD mice. _Neuron_ 64, 828–840 (2009). Article CAS PubMed PubMed Central MATH Google Scholar * DeGuire, S. M. et al. N-terminal Huntingtin (Htt)
phosphorylation is a molecular switch regulating Htt aggregation, helical conformation, internalization, and nuclear targeting. _J. Biol. Chem._ 293, 18540–18558 (2018). Article CAS PubMed
PubMed Central MATH Google Scholar * Isas, J. M., Langen, A., Isas, M. C., Pandey, N. K. & Siemer, A. B. Formation and Structure of Wild Type Huntingtin Exon-1 Fibrils.
_Biochemistry_ 56, 3579–3586 (2017). Article CAS PubMed Google Scholar * Phan, T. T. & Schmit, J. D. Thermodynamics of huntingtin aggregation. _Biophys. J._ 118, 2989–2996 (2020).
Article ADS CAS PubMed PubMed Central MATH Google Scholar * Nekooki-Machida, Y. et al. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different
cytotoxicity. _Proc. Natl. Acad. Sci._ 106, 9679–9684 (2009). Article ADS CAS PubMed PubMed Central MATH Google Scholar * Jain, G. et al. Inhibitor-based modulation of huntingtin
aggregation reduces fibril toxicity. _bioRxiv_ preprint https://doi.org/10.1101/2023.04.24.537565 (2023). * Tam, S. et al. The chaperonin TRiC blocks a huntingtin sequence element that
promotes the conformational switch to aggregation. _Nat. Struct. Mol. Biol._ 16, 1279–1285 (2009). Article CAS PubMed PubMed Central MATH Google Scholar * Michalek, M., Salnikov, E.
S., Werten, S. & Bechinger, B. Membrane interactions of the amphipathic amino terminus of huntingtin. _Biochemistry_ 52, 847–858 (2013). Article CAS PubMed Google Scholar * Ceccon,
A. et al. Interaction of Huntingtin exon-1 peptides with lipid-based micellar nanoparticles probed by solution NMR and Q-band pulsed EPR. _J. Am. Chem. Soc._ 140, 6199–6202 (2018). Article
CAS PubMed PubMed Central MATH Google Scholar * Gasteiger, E. et al. ExPASy: the proteomics server for in-depth protein knowledge and analysis. _Nucleic Acids Res._ 31, 3784–3788
(2003). Article CAS PubMed PubMed Central MATH Google Scholar * Mandal, A., Boatz, J. C., Wheeler, T. B. & van der Wel, P. C. A. On the use of ultracentrifugal devices for routine
sample preparation in biomolecular magic-angle-spinning NMR. _J. Biomol. NMR_ 67, 165–178 (2017). Article CAS PubMed PubMed Central Google Scholar * Stevens, T. J. et al. A software
framework for analysing solid-state MAS NMR data. _J. Biomol. NMR_ 51, 437–447 (2011). Article CAS PubMed PubMed Central MATH Google Scholar * Bennett, A. E., Rienstra, C. M., Auger,
M., Lakshmi, K. & Griffin, R. G. Heteronuclear decoupling in rotating solids. _J. Chem. Phys._ 103, 6951–6958 (1995). Article ADS CAS Google Scholar * Takegoshi, K., Nakamura, S.
& Terao, T. 13C–1H dipolar-assisted rotational resonance in magic-angle spinning NMR. _Chem. Phys. Lett._ 344, 631–637 (2001). Article ADS CAS MATH Google Scholar * Baldus, M. &
Meier, B. H. Total correlation spectroscopy in the solid state. The use of scalar couplings to determine the through-bond connectivity. _J. Magn. Reson., Ser. A_ 121, 65–69 (1996). Article
ADS CAS MATH Google Scholar * Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. _SoftwareX_ 1, 19–25
(2015). Article ADS MATH Google Scholar * Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. _J. Chem. Phys._ 126, 014101 (2007). Article ADS
PubMed Google Scholar * Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. _J. Appl. Phys._ 52, 7182–7190 (1981). Article ADS
CAS MATH Google Scholar * Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An _N_·log(_N_) method for Ewald sums in large systems. _J. Chem. Phys._ 98, 10089–10092 (1993).
Article ADS CAS MATH Google Scholar * Essmann, U. et al. A smooth particle mesh Ewald method. _J. Chem. Phys._ 103, 8577–8593 (1995). Article ADS CAS MATH Google Scholar * Hess, B.
P-LINCS: A parallel linear constraint solver for molecular simulation. _J. Chem. Theory Comput._ 4, 116–122 (2008). Article CAS PubMed MATH Google Scholar * Bagherpoor Helabad, M., van
der Wel, P. C. A. & Miettinen, M. S. Integrative determination of atomic structure of mutant huntingtin exon 1 fibrils implicated in huntington disease — data files. _Zenodo_ repository
https://doi.org/10.5281/zenodo.13926360 (2024). * Maier, J. A. et al. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. _J. Chem. Theory Comput._ 11,
3696–3713 (2015). Article CAS PubMed PubMed Central MATH Google Scholar * Robertson, M. J., Tirado-Rives, J. & Jorgensen, W. L. Improved peptide and protein torsional energetics
with the OPLS-AA force field. _J. Chem. Theory Comput._ 11, 3499–3509 (2015). Article CAS PubMed PubMed Central MATH Google Scholar * Huang, J. et al. CHARMM36m: an improved force
field for folded and intrinsically disordered proteins. _Nat. Methods_ 14, 71–73 (2017). Article CAS PubMed MATH Google Scholar * Jorgensen, W. L., Chandrasekhar, J., Madura, J. D.,
Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. _J. Chem. Phys._ 79, 926–935 (1983). Article ADS CAS Google Scholar * Kuhlman, B.,
O’Neill, J. W., Kim, D. E., Zhang, K. Y. & Baker, D. Accurate computer-based design of a new backbone conformation in the second turn of protein L. _J. Mol. Biol._ 315, 471–477 (2002).
Article CAS PubMed Google Scholar * Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. _J. Mol. Graph._ 14, 33–38 (1996). Article CAS PubMed Google Scholar *
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. _Protein Sci._ 32, e4792 (2023). Article CAS PubMed PubMed Central Google Scholar * Kim, J. U. &
O’Shaughnessy, B. Nanoinclusions in dry polymer brushes. _Macromolecules_ 39, 413–425 (2006). Article ADS CAS MATH Google Scholar * Yaneva, J., Dimitrov, D. I., Milchev, A. &
Binder, K. Nanoinclusions in polymer brushes with explicit solvent — A molecular dynamics investigation. _J. Colloid Interface Sci._ 336, 51–58 (2009). Article ADS CAS PubMed MATH
Google Scholar * Hofmann, H. et al. Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. _Proc. Natl. Acad. Sci._ 109,
16155–16160 (2012). Article ADS CAS PubMed PubMed Central MATH Google Scholar Download references ACKNOWLEDGEMENTS This study was supported by funds from the CHDI foundation contract
A-17778 (P.v.d.W., M.M.), the CampagneTeam Huntington (P.v.d.W.) and prior funding via NIGMS R01 GM112678 (P.v.d.W.). Part of the work has been performed under the Project HPC-EUROPA3
(INFRAIA-2016-1-730897; M.B.H.), with the support of the EC Research Innovation Action under the H2020 Program; in particular, M.B.H. gratefully acknowledges the support of Zernike Institute
for Advanced Materials of the University of Groningen and the computer resources and technical support provided by SURF HPC. Majority of the calculations presented here were carried out on
the MPG supercomputers RAVEN and COBRA hosted at MPCDF. Financial support by the Volkswagen Foundation (86110; M.S.M.) and by the Trond Mohn Foundation (BFS2017TMT01; M.S.M.) is gratefully
acknowledged. This work benefited from access to the uNMR-NL facility at Utrecht University, an Instruct-ERIC center, with financial support provided by Instruct-ERIC (PID 18439; P.v.d.W.).
We thank Dr. Alessia Lasorsa for recording some of the ssNMR experiments. M.B.H. thanks Dr. Matthias Elgeti for his support during the writing of this manuscript and for his valuable
feedback. FUNDING Open Access funding enabled and organized by Projekt DEAL. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Theory and Bio-Systems, Max Planck Institute of
Colloids and Interfaces, 14476, Potsdam, Germany Mahdi Bagherpoor Helabad & Markus S. Miettinen * Institute for Drug Discovery, Leipzig University Medical Center, 04103, Leipzig, Germany
Mahdi Bagherpoor Helabad * Institute of Chemistry, Martin Luther-University Halle-Wittenberg, 06120, Halle (Saale), Germany Mahdi Bagherpoor Helabad * Zernike Institute for Advanced
Materials, University of Groningen, 9747 AG, Groningen, The Netherlands Irina Matlahov, Greeshma Jain & Patrick C. A. van der Wel * NMR Spectroscopy, Bijvoet Centre for Biomolecular
Research, Department of Chemistry, University of Utrecht, 3584 CH, Utrecht, The Netherlands Raj Kumar & Markus Weingarth * Fachbereich Physik, Freie Universität Berlin, 14195, Berlin,
Germany Jan O. Daldrop & Markus S. Miettinen * Department of Chemistry, University of Bergen, 5007, Bergen, Norway Markus S. Miettinen * Computational Biology Unit, Department of
Informatics, University of Bergen, 5008, Bergen, Norway Markus S. Miettinen Authors * Mahdi Bagherpoor Helabad View author publications You can also search for this author inPubMed Google
Scholar * Irina Matlahov View author publications You can also search for this author inPubMed Google Scholar * Raj Kumar View author publications You can also search for this author
inPubMed Google Scholar * Jan O. Daldrop View author publications You can also search for this author inPubMed Google Scholar * Greeshma Jain View author publications You can also search for
this author inPubMed Google Scholar * Markus Weingarth View author publications You can also search for this author inPubMed Google Scholar * Patrick C. A. van der Wel View author
publications You can also search for this author inPubMed Google Scholar * Markus S. Miettinen View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS M.B.H. and J.O.D. performed and analysed MD simulations; M.B.H., J.O.D., and M.S.M. planned and designed the simulations; I.M., R.K, and G.J. performed and analysed NMR
experiments; I.M. and G.J. prepared fibril samples; I.M., R.K., G.J., M.W., and P.C.A.v.d.W. designed experiments; M.B.H., I.M., P.C.A.v.d.W., and M.S.M. wrote the paper. All authors
reviewed and edited the manuscript and approved its final form. CORRESPONDING AUTHORS Correspondence to Patrick C. A. van der Wel or Markus S. Miettinen. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewers for their contribution to the peer review of
this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY MOVIE 1 REPORTING SUMMARY TRANSPARENT PEER REVIEW FILE RIGHTS
AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in
any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Bagherpoor
Helabad, M., Matlahov, I., Kumar, R. _et al._ Integrative determination of atomic structure of mutant huntingtin exon 1 fibrils implicated in Huntington disease. _Nat Commun_ 15, 10793
(2024). https://doi.org/10.1038/s41467-024-55062-8 Download citation * Received: 21 October 2023 * Accepted: 29 November 2024 * Published: 30 December 2024 * DOI:
https://doi.org/10.1038/s41467-024-55062-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Mariah Carey accidentally flashes NIPPLES as she suffers wardrobe fail in sheer mini dressWe use your sign-up to provide content in ways you've consented to and to improve our understanding of you. This may inc...
Hong kong's diy artist christina li takes listeners on a journey through her musical developmentHong Kong-based singer-songwriter, producer and arranger Christina Li is a prime example of an up-and-coming DIY recordi...
Albanvale - 3021 - location - abc newsThis service may include material from Agence France-Presse (AFP), APTN, Reuters, AAP, CNN and the BBC World Service whi...
Kerala state lottery win win w-523 result out; winners’ list hereThe Kerala State Lottery department has announced the results for Win Win W-523 lottery on their official website today ...
Stalin’s bomb: how the soviet union went nuclear | thearticleHow did the Soviet Union under Stalin first manage to create nuclear weapons? I shall first briefly review the scientifi...
Latests News
Integrative determination of atomic structure of mutant huntingtin exon 1 fibrils implicated in huntington diseaseABSTRACT Neurodegeneration in Huntington’s disease (HD) is accompanied by the aggregation of fragments of the mutant hun...
Bed bugs: three 'natural treatments' to get rid of the pestsOnce bed bugs have settled into your bed, mattress, headboard, or side cabinet, the small insects can be difficult to ge...
How to live longer: add these foods and vitamins to your dietHolly Zoccolan is the founder of online wellness community, The Health Zoc. She aims to help others make informed choice...
Fatty liver disease: swelling is a symptomIt is unclear exactly what causes the build-up of fat seen in NAFLD but it's usually seen in people who are overwei...
The page you were looking for doesn't exist.You may have mistyped the address or the page may have moved.By proceeding, you agree to our Terms & Conditions and our ...