Lowering of the singlet-triplet energy gap via intramolecular exciton-exciton coupling
Lowering of the singlet-triplet energy gap via intramolecular exciton-exciton coupling"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Download PDF Article Open access Published: 08 October 2024 Lowering of the singlet-triplet energy gap via intramolecular exciton-exciton coupling Clara Schäfer ORCID:
orcid.org/0000-0001-7634-39801 na1, Rasmus Ringström ORCID: orcid.org/0000-0002-3340-957X2 na1, Jörg Hanrieder ORCID: orcid.org/0000-0001-6059-198X3,4, Martin Rahm ORCID:
orcid.org/0000-0001-7645-59232, Bo Albinsson ORCID: orcid.org/0000-0002-5991-78632 & …Karl Börjesson ORCID: orcid.org/0000-0001-8533-201X1 Show authors Nature Communications volume 15,
Article number: 8705 (2024) Cite this article
6791 Accesses
4 Citations
7 Altmetric
Metrics details
Subjects Excited statesPhotocatalysisOptical spectroscopy AbstractOrganic dyes typically have electronically excited states of both singlet and triplet multiplicity. Controlling the energy difference between these states is a key factor for making
efficient organic light emitting diodes and triplet sensitizers, which fulfill essential functions in chemistry, physics, and medicine. Here, we propose a strategy to shift the singlet
excited state of a known sensitizer to lower energies without shifting the energy of the triplet state, thus without compromising the ability of the sensitizer to do work. We covalently
connect two to four sensitizers in such a way that their transition dipole moments are aligned in a head-to-tail fashion, but, through steric encumbrance, the delocalization is minimized
between each moiety. Exciton coupling between the singlet excited states considerably lowers the first excited singlet state energy. However, the energy of the lowest triplet excited state
is unperturbed because the exciton coupling strength depends on the magnitude of the transition dipole moments, which for triplets are very small. We expect that the presented strategy of
designed intramolecular exciton coupling will be a useful concept in the design of both photosensitizers and emitters for organic light emitting diodes as both benefits from a small
singlet-triplet energy gap.
Similar content being viewed by others Highly efficient luminescence from space-confined charge-transfer emitters Article 15 June 2020 Hybridization ofshort-range and long-range charge transfer excited states in multiple resonance emitter Article Open access 09 August 2023 Towards efficient near-infrared fluorescent organic light-emitting
diodes Article Open access 21 January 2021 Introduction
Triplet photosensitizers (PS) are crucial for photocatalysis1,2,3,4, triplet-triplet annihilation upconversion5,6,7, photodynamic therapy8,9,10,11, and various other fields and applications.
New avenues of utilizing PS are frequently discovered and with the growing interest, the development of organic, highly efficient PS has gained more attention. The crucial property of a
triplet PS is its rate of intersystem crossing (ISC). ISC is a spin forbidden process which can be realized by magnetic perturbations, like spin-orbit coupling12. To achieve a high rate of
ISC, traditional PS make use of the heavy atom effect, known to enhance spin-orbit coupling13,14. Heavy elements like Pt, Pd, Ru, Ir, Se, I and Br have shown to enhance ISC15,16. However,
there are considerable disadvantages of using heavy atoms to increase the rate of ISC, such as their scarceness, toxicity, and cost. Furthermore, the increased spin-orbit coupling also
reduces the triplet state lifetime, which is a negative feature when used in PS applications17. To avoid some of these drawbacks, development and improvement of organic, heavy atom free
triplet PS is in high demand.
Several strategies have been developed to realize an enhanced rate of ISC for compounds without incorporating heavy atoms. One that has gained recognition is spin-orbit charge transfer
intersystem crossing (SOCT-ISC)9,18,19. Systems exhibiting this phenomenon are usually donor-acceptor dyads able to perform intramolecular excited state electron transfer reactions. Upon
photoinduced electron transfer, the system forms a charge separated (CS) state. The donor and acceptor moieties are designed to have an orthogonal geometry towards each other. Due to the
orthogonality of the system, the change in molecular orbital angular momentum compensates for the change in spin angular momentum, enhancing the rate of ISC20. Thus, from the CS state the
molecule can undergo charge recombination to the triplet state in an efficient manner. This approach does not rely on the heavy atom effect and is therefore highly appropriate for the design
of heavy atom free photosensitizers.
Recently, several triplet photosensitizers undergoing SOCT-ISC based on the BODIPY scaffold have been reported5,8,21,22,23. Within the scope of these reports, a BODIPY-anthracene dyad stands
out with a high yield of ISC24,25,26. These studies also highlight the importance of the energy alignment between the S1 and CS states. The energy of the CS state is strongly dependent on
the polarity of the solvent, and the S1-CS energy alignment can therefore be tuned by the choice of solvent. In a non-polar solvent (toluene), the CS state was at a higher energy than the S1
state, resulting in a neglectable ISC but a high yield of fluorescence (ФFluo = 0.81). However, with increasing solvent polarity, the CS state is stabilized, resulting in exergonic excited
state electron transfer. The yield of ISC thus increased (ФISC = 0.92) at the expense of the fluorescence (ФFluo = 0.002)24.
Organic dyes such as BODIPY’s have a relatively large S1-T1 energy gap. In all applications relying on a triplet sensitizer, this gap can be regarded as an energy loss. As small an energy
gap as possible is thus preferable. In recent years, the field of organic electronics has been focusing on reducing this energy gap by separating the HOMO and LUMO orbitals in space as doing
so affects the electron exchange interaction12,27,28,29. However, there are other possible mechanisms that theoretically can perturb the S1-T1 energy alignment30,31. One of these is exciton
coupling, the effect seen in J-aggregates32. Exciton coupling influences the singlet state energy without affecting either the triplet or CS state energies significantly. Even before the
development of the theory of exciton coupling it was observed that aggregation affects the fluorescence and phosphorescence differently33. Curiosity about this observation amongst other
things, later lead to the finding that exciton coupling influences the singlet state energy without affecting either the triplet or CS state energies significantly34. This is because the
effect is based on a Columbic coupling of the transition dipole moments associated with the respective states, and only singlet states have an appreciable magnitude of their transition
dipole moments35. Thus, from a theoretical perspective, assembling an organic PS in the form of a J-aggregate, will reduce energy losses by allowing a red-shifted excitation wavelength.
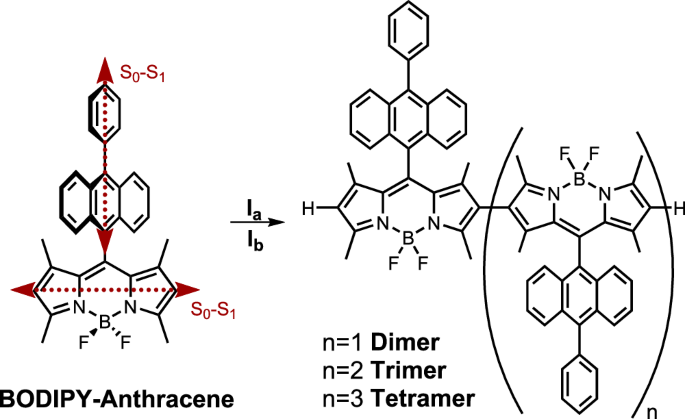
Here, BODIPY-anthracene dyads were β-tethered into oligomers (2–4 units) in a non-conjugated fashion. The β-tethering aligns the S0-S1 transition dipole moments of the BODIPYs in a
‘head-to-tail’ fashion. Intramolecular exciton coupling thus occurs, giving an allowed low energy transition that depends on the number of coupled BODIPY-anthracene moieties. This is the
same effect as seen in J-aggregates and has also previously been observed in ethylene-bridged BODIPY oligomers36,37. The β-tethered oligomers have almost unperturbed energies of their T1 and
CS states because direct electronic communication between the moieties was designed to be suppressed. However, they are still able to perform ISC. The formation of covalently tethered
J-aggregate mimics is thus a viable path to reduce energy losses in organic sensitizers. This by selectively lowering the S1 state without significantly influencing the energy of the triplet
state.
ResultsTo lower the S1 energy without affecting the CS and T1 energies, it is important to align the S0-S1 transition dipole moments without causing an extension of the aromatic system in
oligomers. This can be achieved if steric hindrance forces the individual moieties into an orthogonal geometry. The BODIPY anthracene dyad was synthesized following the literature
procedure38. Dimerization of BODIPY dyes has previously been achieved in the meso, α, and β positions39,40,41. Of these attachment positions, β-tethering also leads to oligomerization of
BODIPY dyes42,43,44. Both conjugated oligomers42 as well as non-conjugated oligomers, where methyl groups on adjacent carbons result in a large dihedral angle between moieties due to steric
hindrance43,44, have been accessed. The conjugated BODIPY oligomers were obtained via Suzuki-Miyaura coupling. The benefit of this reaction is the controllability of the degree of
oligomerization. However, a fully conjugated system would not just influence the singlet energy, but also the triplet energy to a similar extent. The non-conjugated systems were obtained by
treating the monomer with either the hypervalent iodine reagent phenyliodine(III)bistrifluoroacetate (PIFA), or anhydrous iron(III)trichloride (Fig. 1). In both cases, trimers were the
highest order of oligomers isolated43,44. However, polymerization was achieved using FeCl3, when leaving the reaction for a longer time. Both conditions were tested here, resulting in the
dimer regardless of conditions used (Supplementary Note 1, 2, Supplementary Figs. 1–19, Supplementary Tables 1, 2). The reaction using FeCl3 yielded the dimer to a larger extent, 17% as
compared to 8% when using PIFA. Furthermore, the reaction using FeCl3 also yielded higher oligomers, the trimer, and the tetramer. Atropisomers of the trimer could be isolated. These are
hypothesized to correspond to the cis and trans isomers with regards to the anthracene units on the end BODIPYs and show discernible 1H NMR spectra (Supplementary Figs. 11, 14;
unfortunately, the distance in between the anthracene units is too large to obtain NOESY signals, which would allow for identification of the two different isomers). After purification, only
trace amounts, 1–2% of the two atropisomers of the trimer, and >1% of the tetramer were obtained. However, the small amounts collected were enough to confirm their structure through 1H, 13C
(Supplementary Dataset 1), diffusion NMR and MALDI, and to probe the excited state energetics and dynamics.
Fig. 1: The BODIPY-anthracene dyad and its oligomers.Synthesis of the meso-10-phenylanthracene-BODIPY dimer (n = 1), trimer (n = 2) and tetramer (n = 3). Reaction conditions: (Ia) PIFA, BF3·OEt2, DCM, −78 °C - r.t, 1 h. (Ib) anhydrous FeCl3,
DCM, r.t., 25 min. The dimer was obtained using Ia and Ib. The trimer and tetramer were obtained using Ib. The S0-S1 transition dipole moments for the anthracene and BODIPY units are
depicted as red, dashed arrows24.
Full size imageThe energies of the singlet and triplet statesIn the presentation of the excited state energetics, we start by discussing the S1 state. We then continue with the T1 and the CS states, so that we in the conclusion can discuss the
complete energetic picture of our systems. To obtain information on the S1 state energy, and to see if it could be decreased successfully, the absorption spectra of the oligomers were
analyzed. The two conformers of the trimer exhibit identical photophysical properties. In this discussion, the results are therefore combined as the trimer. The absorption envelope (Fig. 2a)
of all compounds can be deconvoluted into an anthracene part (370–400 nm) and a BODIPY part (500–600 nm). The wavelength (i.e., energy) of the anthracene absorption is not affected by
oligomerization. This means that the anthracene moieties are located far enough apart, and/or the orientation between them is disadvantageous, for Columbic interactions between the
transition dipole moments to become significant. The situation for the BODIPY part, where the transition dipole moments are designed to be in a head-to-tail fashion, is quite different. The
absorption of the oligomers is shifted to longer wavelengths, with absorption maxima at 506, 542, 569, and 583 nm for the monomer, dimer, trimer, and tetramer, respectively. As expected, the
absorption energy converges as the number of moieties increases. The excitation energy thus decreases asymptotically, reducing the gain in making even longer oligomers (Fig. 2b).
Furthermore, the magnitude of the transition dipole moments were calculated from the molar absorptivity spectra (Supplementary Note 3, Supplementary Fig. 20)45. As expected for an exciton
coupled system, the transition dipole moments scale well with the square root of the number of moieties (Fig. 2c)35, although limited purity of the tetramer (Supplementary Fig. 17) is
exhibited as a less than expected transition dipole moment for that molecule.
Fig. 2: Energetics of excited states.a Molar absorptivity spectra of the synthesized oligomer series recorded in DCM. b The relative energy of the absorption maximum versus number of BODIPY units in the oligomers. Experimental
values (circles) are compared to theoretical values based on a point dipole interaction (squares) and DFT (triangles, solid line for S1 and dashed line for T1). c Transition dipole moments
(TDMs) versus number of BODIPY units in the oligomers. Experimental values (circles) are compared to DFT calculations (triangles), and the exciton coupling model (squares) where the
magnitude of the monomer transition dipole moment is scaled with the square root of the number of monomer units. Source data are provided as a Source Data file.
Full size imagePreviously reported conjugated BODIPY dimers and trimers, show a different trend than we observe here. Much larger bathochromic shifts have been reported for conjugated oligomers42. The
different observations indicate different main communication pathways for conjugated and non-conjugated oligomers. In the latter, the lack of conjugation in between the monomer units
minimizes electron delocalization. Exciton coupling could however explain the observed spectral changes, as it does not involve conjugation between moieties46. Using a point dipole
approximation, the Coulombic interaction energies between moieties in the oligomers were calculated (Supplementary Note 4). Based on this, the spectral shift solely due to Coulombic
interactions could be extracted. The vast majority of the experimentally observed spectral shift can be reproduced by taking Coulombic interactions into account. Thus, the observed spectral
changes are mainly a consequence of exciton coupling rather than electronic delocalization.
Quantum mechanical calculations were performed to validate the degree to which the triplet energy remains constant across the series. The geometry of singlet and triplet ground states were
optimized in vacuum at the ωB97XD/6-31 g(d) level of theory (Supplementary Fig. 21, Supplementary Dataset 2) and singlet excitation energies were obtained through TD-DFT using the singlet
ground state geometry using Gaussian 16, revision B.0147,48. No imaginary frequencies for the optimized ground state geometry verifies that a minimized structure was obtained. Although the
overall trend is captured well, these calculations overestimate the reduction of the S1 energy as well as the increase of the transition dipole moment with oligomer length (Fig. 2b, c,
Supplementary Table 3). But, more importantly, the calculations predict that the energy of the T1 states only marginally decrease across the monomer-to-tetramer series (Fig. 2b).
Emissionefficiency and dynamics
We will now turn our attention to the energy relaxation pathways towards the charge separated state after exciting the oligomers. Figure 3 shows the steady state emission spectra of the
monomer and oligomers in toluene (for other solvents and oligomers, see Supplementary Figs. 22–26). Clear BODIPY like emission envelopes that follow the same energetic trend as the
absorption spectra can be seen. However, when increasing the polarity of the solvent, a distinguishable second band is observed. This has, for the monomer, previously been ascribed to
emission from the charge separated state24. The emission from the CS state is intense and spectrally well resolved for the monomer, but it is also present to a minor degree in the oligomers
as a shoulder at lower energies in the high polar solvents (Fig. 3).
Fig. 3: Emission from monomer and oligomers.Emission of the monomer (a), dimer (b), trimer (d) and tetramer (e) are shown in toluene, DCM and ACN. Emission of all oligomers in toluene are shown in (c). The vertical lines show the
energy of the CS state in DCM (blue) and ACN (green) estimated from electrochemistry. Source data are provided as a Source Data file.
Full size imageIt is difficult to draw any conclusions regarding the efficiency of reaching the CS state based on the magnitude of the CS emission. It is better to compare how well the presence of an
energetically accessible CS state quenches the fluorescence (Table 1). The fluorescence quantum yield of the monomer shows a drop from 0.80 in toluene to 0.02 in DCM and 0.01 in ACN. This
effect can be explained by the polarity and thus the dielectric constants of the solvents. The Rehm-Weller equation predicts that the energy of the CS state lowers as the dielectric constant
increases15,49. ACN has a larger dielectric constant than DCM and toluene. Therefore, the CS state in ACN is lower than in DCM, which is lower than in toluene. The driving force for
populating the CS state is thus larger in ACN and DCM, explaining the rise of the second emission band in these solvents. The drop in ФEm is less intense for the dimer, trimer and tetramer
in DCM, but it is still considerable in ACN. From a driving force perspective, this difference indicates that the energy of the singlet state decreases more than the CS state in the
oligomers. Thus, the energy difference between the S1 and CS states only remains large enough in ACN for charge separation to outcompete fluorescence in the longer oligomers.
Table 1Spectroscopic data of the BODIPY oligomers collected in different solventsFull size table
To evaluate the efficiency of internal conversion from higher excited states towards the S1 state, different excitation energies were used to excite the monomer. Excitation of the BODIPY
moiety is described above. Upon excitation at higher energies, at the anthracene absorption, the emission spectrum looks almost identical (Supplementary Fig. 22). The only difference is seen
in the relative emission intensity between the S1 and the CS states emission bands. When the anthracene unit is excited, the CS state emission is relatively increased. This indicates that
charge separation competes with Sn to S1 internal conversion in these systems.
The fluorescence quantum yield of the monomer is greatly reduced in ACN compared to toluene. However, the observed emission lifetime is not reduced to the same extent (Table 1, see
Supplementary Figs. 27–33 for emission decays). This surprising feature has been previously observed24, and we will try to rationalize it. When recording the emission lifetime for the
monomer, a long-lived transient as well as an instrument response function limited signal is present when monitoring the S1 but not the CS state emission (Supplementary Figs. 27, 29). The
rate of charge separation is thus faster than the time resolution of our instrumentation. The long-lived emission must therefore not be due to the directly excited BODIPY unit. Furthermore,
when observing the steady state emission as a function of decreasing temperature (Supplementary Fig. 34), we see a reduction of the S1 emission until a plateau value is reached. Based on
these two observations, we suggest that the plateau value represent prompt emission from the S1 state, and the temperature dependent emission represents endothermic charge recombination from
the CS state back to the S1 state. To strengthen this hypothesis, the system was modeled using rate equations (Supplementary Note 5). In the modeling, it was assumed that the S1 and CS
states are connected by a microscopic reversibility, implying that the ratio of the rate constants between them being equal to the Boltzmann factor. The modeled system thus strongly
resembles that used when modeling molecules exhibiting E-type delayed fluorescence (with the triplet state then being replaced by a CS state). Using the energy difference between the two
states as a fitting parameter, both the steady state and time resolved temperature dependent emission could be qualitatively reproduced (Supplementary Fig. 35). Furthermore, the fitted
energy difference between the S1 and CS states was 12 kJ/mol (this corresponds to a 28 nm shift compared to the S1 energy), which is well in line with the emission data (Fig. 3). It is thus
very plausible that the observed long lifetime represents the lifetime of the CS state, illustrating the complex dynamics in these systems.
Energy of the CS stateTo investigate whether the energy of the CS state is influenced by oligomerization, the redox potentials of the monomer and the dimer were determined in DCM (Supplementary Note 6). Limited
solubility in ACN prevented experimental determination of the CS state energy, and we therefore relied on the Rehm-Weller equation for this value. The monomer shows a reversible one-electron
oxidation and reduction at +0.71 V and −1.73 V vs. Fc/Fc+, respectively in DCM (Supplementary Fig. 36a, Supplementary Table 4). These values correlate very well with the previously reported
redox potentials of the monomer24, where the reduction was assigned to the BODIPY and oxidation to the anthracene moieties. The dimer in DCM shows two reversible one-electron waves for
reduction (Supplementary Fig. 36b, Supplementary Table 4). The first reduction signal at −1.71 V is almost identical to the one observed for the monomer. This agreement indicates that the
reduction of the monomeric unit of the dimer is almost identical to that in the monomer. In contrast to the reduction, only one oxidation potential for the dimer was observed close to the
end of the solvent window. A small shift of 80 meV for the oxidation potential was seen. It is unlikely that this is caused by anthracene-anthracene interactions, because of their large
distance to each other. However, the oxidation peak is not well resolved, and the error margins are therefore large. Nevertheless, the change in S1 energy is notably larger compared to the
change in CS energy (about 160 meV vs 100 meV), and the CS energy change due to oligomerization is small compared to the effect of solvent polarity. It should be noted here that limited
solubility of the trimer and tetramer prevented cyclic voltammetry to be conducted.
It is interesting to compare the energy of the CS state measured electrochemically and spectroscopically. The energy of the electrochemically determined CS state is summarized in
Supplementary Table 5 and shown schematically in Fig. 3 as a vertical line for the monomer and dimer. It is located at a higher energy as compared to the CS state emission maximum, which is
reasonable. Thus, the electrochemically and spectroscopically measured energy of the CS state corroborate each other. In summary, the energy of the CS state is less affected in comparison to
the changes seen for the S1 state by oligomerization. Furthermore, it can be tuned separately by the solvent polarity.
Formation of the triplet stateThe ability of the monomer, dimer, trimer, and tetramer to relax from the CS state to the T1 state was assessed by nanosecond transient absorption. The monomer is known to form the triplet
state in high yield in both DCM and ACN. Indeed, after excitation to the S1 state, all compounds show both a ground state bleach, as well as an excited state absorption in degassed DCM as
well as ACN solutions (Fig. 4a, b). Furthermore, as oxygen is added, all transient absorption features disappear (Supplementary Figs. 37–41). This indicates that all the oligomers populate
the triplet state in these solvents. The kinetics of the recovery to the ground state was fitted well assuming first order kinetics, resulting in lifetimes of around 150 μs for the monomer
and generally longer for the oligomers (Table 1, Fig. 4c, d). The triplet lifetime is on the same order of magnitude as iodinated BODIPY50, and several metal porphyrins. However, what stand
out is that the triplet lifetime does not decrease with the number of moieties in the series, despite that the S1 energy decreases. If the reduction of the singlet energy also reduced the
triplet energy, a reduction of the triplet lifetime would have been expected from the energy gap law51,52,53. Thus, this feature can be explained by the change in singlet energy being
decoupled from the triplet energy.
Fig. 4: Nanosecond transient absorption.Normalized transient absorption spectra of the investigated compounds in (a) DCM and (b) ACN at a time delay of 1 µs obtained with an excitation wavelength corresponding to the absorption
maximum of each of the compounds. c, d Show the ground state bleach kinetics of the monomer, dimer, trimer and tetramer probed at the respective ground state bleach maxima in DCM and ACN,
respectively. Limited solubility of the trimer prevented quantification of ФT in ACN. Source data are provided as a Source Data file.
Full size imageThe yield of intersystem crossing for the monomer is known, and it was therefore used as a reference when determining the yield of intersystem crossing for the oligomers. Figures 4c, d show
decays in DCM and ACN, respectively, using absorption and excitation fluence matched conditions (low solubility of the trimer prevented such experiment in ACN). Under such conditions, the
yield of intersystem crossing can be determined by comparing the ground state bleach shortly after the excitation pulse. In DCM, the yield of intersystem crossing reduces through the series
(Table 1). This is an expected result as the driving force for forming the CS state is continuously reduced through the series. Still, it is rewarding to simulate the yields of emission and
intersystem crossing from known measurables in order to gain a thorough understanding of the observed photophysics in the series. A rate equation model was therefore constructed
(Supplementary Note 7). It is based on a three-level system, the S1, CS, and T1 states, of which the S1 and CS states are in dynamic equilibrium with each other (Fig. 5a), where the
individual rate constants are shown in Supplementary Note 7 and Supplementary Tables 6, 7.
Fig. 5: Simulating the photophyiscs with rate equations.a The kinetic model used to simulate the photophysics of the oligomers. Note that SOCT-ISC entails a direct conversion of the CS state into the T1 state, involving a reverse electron
transfer in conjunction with a spin inversion process. b, c Simulated (circles) and experimental (triangles) quantum yields of fluorescence (black) and ISC (blue) in DCM (b) and ACN (c).
Source data are provided as a Source Data file.
Full size imageFigure 5b shows the simulated and measured quantum yields of fluorescence and intersystem crossing in DCM. The trends in oligomer lengths are captured very well, with a first increasing and
then decreasing yield of fluorescence, and a continuously decreasing yield of intersystem crossing. The dependence of the fluorescence yield on oligomer length can be explained by the
balance between a decreased driving force for charge separation and an increased non-radiative decay to the ground state. The energetics of the charge separation goes from exothermic for the
monomer to endothermic for the oligomers. The result being that the equilibrium concentrations are shifted towards the S1 state with an increased number of moieties in the oligomer. The
energetics also explain why a very fast decay component in the TCSPC experiments only could be seen for the monomer (vide supra). Counteracting the shift in the dynamic equilibrium
concentration is the non-radiative decay to the ground-state, which increases throughout the series. That internal conversion increases with a reduced excitation energy is commonly observed
and is usually explained by the energy gap law51,52. We note, however, that for J-aggregates there are examples where a reduced reorganization energy counteracts the effect of the energy gap
law32,54,55. The simulations also capture the yields of fluorescence and intersystem crossing in ACN solution very well. Here, a high driving force for charge separation results in a low
yield of fluorescence throughout the series. A slight increase in emission yield with oligomer length is observed, which can be explained from a reduction in the driving force with oligomer
length. Overall, the trends of the fluorescence and intersystem crossing yields with oligomer length is quite straightforwardly explained by the driving force for charge separation. Thus,
highlighting the importance of the delicate balance of the energetics between the S1, CS and T1 states for the SOCT-ISC process. Optimization of these energetics could for instance be done
by changing the oxidation potential of the anthracene moiety through chemical modifications or by changing the solvent. To demonstrate that solvent polarity can be used as an optimization
tool to increase the yield of triplet formation, measurements were conducted on the dimer in benzonitrile, a solvent with a polarity (ε = 25.9) between that of DCM and ACN. As expected, the
dimer exhibited an emission quantum yield (29%) that was intermediate between those observed in DCM and ACN. Additionally, the triplet quantum yield (24%; Supplementary Fig. 43), increased
by 41% and 71% compared to DCM and ACN, respectively. Also, although our simulations capture the overall trend well, the absolute values are less well explained. We here note that it is
possible to perform excellent fits with our model, by for instance using the energy of the CS state, the rate of charge recombination to the T1 state, or the rate of charge separa
tion as fitting parameters. However, as we do not know which of these parameters changes most, we stop at the point where approximated rates fit the overall trends in yields well.
The yields of intersystem crossing and fluorescence are relatively low for the oligomers in ACN. This is attributed to an increase in the non-radiative rate constant in ACN for the CS state,
which is well explained by Marcus theory for electron transfer. To investigate the rate changes throughout the series, we conducted femtosecond transient absorption experiments (which
corroborated the time-resolved emission experiments) and analyzed the photophysics using Marcus theory (Supplementary Note 8, Supplementary Figs. 44–46). The initial formation of the CS
state as well as its relaxation to the triplet and ground states are electron transfer processes. The rate of electron transfer reactions normally increase steeply with the driving force
until the driving force equals the reorganization energy (so-called Marcus normal region). With further increase in the driving force, the rate decreases sharply (so-called Marcus inverted
region). Redox potentials for the monomer and dimer could be obtained (vide infra), and these were used to analyse the rate for charge recombination to the triplet and ground states. The
obtained rates align with the predictions from Marcus theory (Supplementary Fig. 46). The rate of recombination to the triplet state increases with increasing driving force, with the monomer
exhibiting a larger driving force and consequently a higher rate. Conversely, the rate of recombination to the ground state decreases with increasing driving force, meaning the monomer,
having a larger driving force, shows a slower recombination rate to the ground state. This combination gives the monomer the prerequisites for forming triplets more efficiently than the
oligomers, explaining the short lifetime observed for the CS state of the oligomers. Thus, the increase in the non-radiative rate with oligomer length is not a consequence of oligomerization
as such, but rather of the mechanism through which the triplet state is reached. Furthermore, the recombination from the CS to the ground state is likely to be in the inverted Marcus regime
for any molecular system. High efficiencies for the SOCT-ISC mechanism are therefore only feasible at considerable CS state energies because the unwanted recombination to the ground state
becomes slower the higher the CS state energy. Thus, limiting this mechanism to energies roughly equaling the visible part of the electromagnetic spectrum. It is important to note that the
low yields, attributed to fast ground state recombination in the oligomers, could have other explanations beyond the one suggested by the analysis based on Marcus theory. However,
investigating these alternative explanations is beyond the scope of this work.
DiscussionsExciton coupling is shown to selectively stabilize the first excited singlet state energy, without significantly perturbing the energy of the first excited triplet state. This approach thus
enables a route to significantly reduce energy losses for triplet sensitizers. We show this in a so-called SOCT-ISC sensitizer by synthesizing a set of new oligomeric BODIPY-anthracene
dyads. The design principles in the coupling of dyad moieties focused on the transition dipole moments being aligned in a head-to-tail fashion, but steric hindrance breaking the conjugated
system. The oligomers show a gradual lowering of the S1 energy with length (Fig. 6). The dimer shows a shift of 36 nm, the trimer 63 nm and the tetramer 77 nm compared to the monomer. This,
as well as the increasing molar absorptivity with oligomer length indicates exciton coupling in the system. Thus, the shift in absorption confirms a successful shift of the S1 state energy
to longer wavelengths.
Fig. 6: A summary of the effect of oligomerization.Schematic energy diagram, illustrating the lowered S1 energy levels of the dimer, trimer and tetramer. The energies of the singlet states were taken as the peak maximum of the absorption
spectra (Fig. 2b). The energies of the charge separated states were estimated from the redox potentials and are shown for the monomer in solid lines and dimer in dotted lines (Supplementary
Table 5). The energies of the triplet states were taken from quantum mechanical calculations (Fig. 2b).
Full size imageThe energy of the charge separated state, where the BODIPY and anthracene units are reduced and oxidized, respectively, is highly dependent on solvent polarity. The choice of solvent
therefore modulates the driving force for charge separation from the S1 state. Because of the lower energy of the S1 state in the oligomers compared to the monomer, a solvent with higher
polarity was needed to quench the fluorescence for the oligomers. The dynamics of the S1-CS state interaction was examined in detail for the monomer. It was found that a model that included
a dynamic equilibrium between these two states were needed to explain the photophysical properties. Recombination from the CS to the ground state was shown to be in the inverted Marcus
regime, practically limiting the SOCT-ISC mechanism to relatively high energies. Electrochemical analysis was used to complement spectroscopical investigation and showed that the reduction
and oxidation potentials of the dimer do not change to a great extent as compared to the monomer.
The ability of all oligomers to populate the triplet state was shown by nanosecond transient absorption spectroscopy, and the experimentally obtained yields could successfully be simulated
by rate equations. Importantly, the corresponding kinetic traces showed no decrease in the triplet lifetime when increasing the oligomer length. That the lifetime of a molecular series does
not reduce with decreasing excitation energy is unusual but can be explained by the triplet energy being constant in the series, which was confirmed by quantum mechanical calculations. The
conserved triplet lifetime highlights a secondary beneficial feature of using exciton coupling for decreasing the excitation energy of a sensitizer. This is important since long triplet
lifetimes are an essential property of photo sensitizers.
We suggest the following design concepts to optimize the photophysical performances beyond those presented herein. Electron delocalization is here restricted through steric bulk. This method
is easily synthetically implemented but allows for a distribution of dihedral angles between monomers (thus a small but variable amount of electron delocalization), resulting in broader
than expected absorption and emission spectral envelopes. By locking the conformation between monomers in an orthogonal orientation, for instance through the introduction of spiro-carbons,
electron delocalization is minimized, and a significant narrowing of the absorbance and emission spectra is expected. Spiro-carbons have previously been introduced in for instance fluorene
to form spirobifluorene, which crystal structure shows perpendicular directions of the two fluorene moieties in the dimer56. The benefits of the SOCT-ISC mechanism are that the lifetime of
the acquired triplet state is long, and that the method works in absence of heavy atoms. The drawback is that it depends on an intermediate CT state, which energy is highly solvent polarity
dependent18. Furthermore, in exciton coupled systems, the driving force for reaching the triplet state is reduced at the same time as the rate of charge recombination to the ground state is
increased, lowering the yield of triplet formation. However, the yields of emission are high and lifetimes relatively long for all oligomers in toluene solution, indicating that exciton
coupling itself does not significantly deactivate the excited state. Thus, limitations for the SOCT-ISC process can most likely be overcome by the utilization of other methods of increasing
ISC, for instance through the incorporation of heavy atoms.
In summary, exciton coupling was used to selectively lower the energy of the S1 state without affecting the triplet state energy to a great extent. This intramolecular exciton-exciton
coupling strategy can be a useful addition in the design of heavy atom free photosensitizers and dyes for light emitting diodes with a small singlet-triplet energy gap.
MethodsSynthesis: All reactions were carried out under ambient conditions unless stated differently, for example performed under N2 atmosphere. Glassware were oven dried prior to use, unless
indicated otherwise. Common reagents, solvents, or materials were obtained from Sigma-Aldrich Chemical Co. and used without further purification. Dry solvents for reactions sensitive to
moisture and/or oxygen were obtained through a solvent purifying system (inert PureSolv-MD-5). Column chromatography was performed using silica gel (VWR 40 to 63 μm) unless stated otherwise.
Preparative thin layer chromatography was performed on silica gel plates (UniplateTM, UV254 indicator, 20×20 cm, 2000 micron, Miles Scientific, purchased from Sigma-Aldrich Chemical Co).
Size exclusion column chromatography (SEC) was performed using Bio-Beads S-X3 Support as a stationary phase and DCM as eluent. NMR spectra (1H, 13C, 19F) were recorded on a BRUKER Avance III
spectrometers (600/700/800 MHz 1H; 151/176/201 MHz 13C; 564/659/753 MHz 19F). The 700 MHz spectrometer is equipped with an AVIII console and a 5 mm QCI H/F–C/N–D–05 Z cryo–probe. The 600
MHz spectrometer is equipped with a NEO console and a QCI H&F/P/C–N–D–05 Z XT cryo–probe. Spectra were taken in CDCl3 (containing tetramethylsilane with 0.00 ppm as an internal reference) or
CD2Cl2 as solvent. Coupling constants (J values) are given in Hertz (Hz) and chemical shifts are reported in parts per million (ppm). 13C spectra are decoupled from 1H. High-resolution MS
was obtained from an Agilent 1290 infinity LC system equipped with an auto sampler in tandem with an Agilent 6520 Accurate Mass Q-TOF LC/MS. LDI MS data were collected with a Rapiflex MALDI
ToF/ToF mass spectrometer (Bruker Daltonics, Bremen, Germany). The instrument is equipped with a Smartbeam 3D UV laser (355 nm) and operated in reflector-positive ionization mode. A number
of 200 shots were acquired at 1 kHz with a mass range set to m/z 400–3200. Melting points were measured using a BÜCHI Melting Point B-545 instrument. IR spectra were recorded using an
INVENIO R FT-IR from BRUKER. Analytical HPLC was performed on a Thermo scientific, Dionex Ultimate 3000 system equipped with a Dionex Ultimate 3000 Variable Wavelength Detector and a Reverse
Phase Dionex Acclaim PolarAdvantage II column. The solvent used was a mixture of MilliQwater and acetonitrile, both containing 0.1% formic acid. The oligomers (Monomer, Dimer, Trimer1,
Trimer2 and Tetramer) were run using the λmax of the regarding compound as the detection wavelength. The compounds were run using a gradient ramping from 5% to 95% MeCN over a time period of
15 min with afterward holding of the final ratio for 10 min in the case of Monomer, dimer, trimer1 and trimer2. The tetramer was run using a gradient ramping from 5% to 95% MeCN over a time
period of 10 min with afterward holding of the final ratio for 20 min. Purities are given in the caption of Supplementary Figs. 2, 6, 11, 14, and 17.
Optical spectroscopy: Absorption spectra were measured using a spectrophotometer (LAMBDA 950, PerkinElmer). Steady-state emission spectra, excitation spectra and emission lifetimes were
measured with a spectrofluorometer (FLS1000, Edinburgh Instrument) and are corrected using the emission correction files provided by the manufacturer. The emission quantum yields were
calculated using the relative method, using a standard (Fluorescein) with known emission quantum yield57. Fluorescein in 0.1 M NaOH (Φf = 0.91) was used as a reference compound for ΦEm
determination57. (Excitation at 480 nm, Refractive index: 1.33). The samples were measured in solution at 22 °C, refractive indices: toluene = 1.497, DCM = 1.4125, ACN = 1.3405. For the
emission lifetime measurements, the samples were excited by a 375 nm or 510 nm picosecond pulsed diode laser (Edinburgh Instruments).
Nanosecond transient absorption measurements were performed on an Edinburgh Instrument LP 980 spectrometer equipped with an ICCD (Andor). A Spectra-Physics Nd:YAG 532 nm laser (pulse width
~7 ns) coupled to a Spectra-Physics primoscan optical parametric oscillator (OPO) was used as pump source. The samples used for transient absorption measurements were prepared in a glovebox
using anhydrous and degassed solvents. The samples used for steady-state absorption and emission spectra were prepared using anhydrous solvents (including molar absorptivities, fluorescence
decays and fluorescence quantum yield). Anhydrous solvents were necessary since the CS state was quenched by the presence of even small amounts of water. There are literature reports showing
that the charge separated state lifetime can be heavily influenced by hydrogen bonding58. However, such interactions are not possible in our system. The quenching mechanism involving water
was not investigated further since it is beyond the scope of this report. In intense laser light the concentration of excited state species might become high enough to allow bimolecular
reactions. For triplet states, triplet-triplet annihilation might occur if two excited molecules collide leading to increased deactivation rate of the excited state population59. The time
dependence of the triplet excited state concentration (if the triplet formation occurs within the instrument response function of the instrument) is given
by:
$$\frac{d\left[{{\scriptstyle{3}\atop}A}{*}\right]}{{dt}}=-2{k}_{{TTA}}{\left[{{\scriptstyle{3}\atop}A}{*}\right]}^{2}-{k}_{T}\left[{{\scriptstyle{3}\atop}A}{*}\right]$$ (1)where kT and kTTA are the first order intrinsic and second order triplet-triplet annihilation rate constants, respectively. Equation 1 has an analytical solution giving the time dependent
triplet excited state concentration.
$$\left[{{\scriptstyle{3}\atop}A}{*}\right]={\left[{{\scriptstyle{3}\atop}A}{*}\right]}_{0}\frac{1-\beta }{{e}^{{k}_{T}t}-\beta },\,{{{\rm{where}}}}\, \beta=\frac{2{k}_{{TTA}}{\left[{{\scriptstyle{3}\atop}A}{*}\right]}_{0}}{{k}_{T}+2{k}_{{TTA}}{\left[{{\scriptstyle{3}\atop}A}{*}\right]}_{0}}$$ (2)
The parameter β describes the ratio of second order decay over total decay rate of the triplet state. Equation 2 was used to fit the nsTA data of the monomer measured at a series of
different intensities in a global fit to obtain the triplet lifetime.
Femtosecond transient absorption (fsTA) measurements were performed with a Ti:sapphire oscillator (Mai-Tai, Spectra Physics) which was used as seed to a regenerative amplifier (Solstice Ace,
Spectra Physics) pumped by a frequency-doubled diode-pumped Nd:YLF laser (Ascend, Spectra Physics). This produced pulses of around 60 fs duration (FWHM) at a 1 kHz repetition rate. The 800
nm output from the amplifier was split, and the two beams were used as pump and probe light. To achieve the desired excitation wavelength of 390 nm (with an energy of 1μJ per pulse at the
sample), we employed an optical parametric amplifier (TOPAS PRIME, Light Conversion Ltd.) The probe light was directed onto a translating CaF2 plate to generate a supercontinuum, while the
pump beam’s timing was adjusted relative to the probe beam using an optical delay stage (with a range from 0 to 10 ns). The supercontinuum was split into a probe and reference beam, and the
probe beam overlapped with the pump at the sample. The transmitted probe and reference beam were directed to optical fibers and detected by a CCD camera (iXon-Andor) operating synchronously
with the 1 kHz laser. The transient spectra were obtained from the difference of the probe light divided by the reference with and without excitation of the sample by the pump beam; 2000
spectra were averaged per delay time using a custom LabVIEW program controlling the setup.
The cyclic voltammetry measurements were performed with a CHI-potentiostat controlled using CHI650A software (version 11.15). Platinum electrodes were used as the working and counter
electrodes and Ag/AgCl in saturated KCl was used as the reference electrode. The measurements were performed in thoroughly degassed (using Argon) dichloromethane (DCM) with 0.1 M
tetra-n-butylammonium perchlorate TBAPF6 (Sigma–Aldrich) at a scan rate of 0.1 V/s. Ferrocene/Ferrocenium (Fc/Fc+) was used as an external standard with E1/2 at approximately 0.61 V vs.
Ag/AgCl in DCM.
Quantum mechanical calculations were performed using Gaussian 16, revision B.0147,48. The geometry of singlet and triplet ground states were optimized in vacuum at the ωB97XD/6-31 g(d) level
of theory and singlet excitation energies were obtained through TD-DFT using the singlet ground state geometry. The energy difference between the ground singlet and triplet states therefore
represents adiabatic transitions, whereas the energy difference between the ground and excited singlet states represents vertical transitions.
Data availabilityThe data that support the findings of this work are available within the Article and its Supplementary Information files. Source data are provided with this paper.
References Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075–10166 (2016).
Article CAS PubMed Google Scholar
Zhao, X., Hou, Y., Liu, L. & Zhao, J. Triplet photosensitizers showing strong absorption of visible light and long-lived triplet excited states and application in photocatalysis: a mini
review. Energy Fuels 35, 18942–18956 (2021).
Article CAS Google Scholar
De Bonfils, P., Péault, L., Nun, P. & Coeffard, V. State of the art of bodipy-based photocatalysts in organic synthesis. Eur. J. Org. Chem. 2021, 1809–1824 (2021).
Article Google Scholar
García-Santos, W. H. et al. Dibromo-BODIPY as an organic photocatalyst for radical–ionic sequences. J. Org. Chem. 86, 16315–16326 (2021).
Article PubMed Google Scholar
Wu, W., Guo, H., Wu, W., Ji, S. & Zhao, J. Organic triplet sensitizer library derived from a single chromophore (BODIPY) with long-lived triplet excited state for triplet–triplet
annihilation based upconversion. J. Org. Chem. 76, 7056–7064 (2011).
Article CAS PubMed Google Scholar
Peng, J., Guo, X., Jiang, X., Zhao, D. & Ma, Y. Developing efficient heavy-atom-free photosensitizers applicable to TTA upconversion in polymer films. Chem. Sci. 7, 1233–1237 (2016).
Article CAS PubMed Google Scholar
Carrod, A. J., Gray, V. & Börjesson, K. Recent advances in triplet–triplet annihilation upconversion and singlet fission, towards solar energy applications. Energy Environ. Sci. 15,
4982–5016 (2022).
Article CAS Google Scholar
Prieto-Montero, R. et al. Exploring BODIPY derivatives as singlet oxygen photosensitizers for PDT. Photochem Photobio. 96, 458–477 (2020).
Article CAS Google Scholar
Nguyen, V.-N., Yan, Y., Zhao, J. & Yoon, J. Heavy-atom-free photosensitizers: from molecular design to applications in the photodynamic therapy of cancer. Acc. Chem. Res. 54, 207–220 (2021).
Article CAS PubMed Google Scholar
Xiao, X., Ye, K., Imran, M. & Zhao, J. Recent development of heavy atom-free triplet photosensitizers for photodynamic therapy. Appl. Sci. 12, 9933 (2022).
Article CAS Google Scholar
Xu, K., Zhao, J., Escudero, D., Mahmood, Z. & Jacquemin, D. Controlling triplet–triplet annihilation upconversion by tuning the PET in aminomethyleneanthracene derivatives. J. Phys. Chem. C.
119, 23801–23812 (2015).
Article CAS Google Scholar
Xu, S. et al. Tuning the singlet-triplet energy gap: a unique approach to efficient photosensitizers with aggregation-induced emission (AIE) characteristics. Chem. Sci. 6, 5824–5830 (2015).
Article CAS PubMed PubMed Central Google Scholar
Balzani V., Ceroni P., Juris A. Photochemistry and photophysics: concepts, research, applications. (John Wiley & Sons, 2014).
Turro N. J., Ramamurthy V., Scaiano J. C. Modern molecular photochemistry of organic molecules, 188. (University Science Books Sausalito, CA, 2010).
Turro N. J., Ramamurthy V., Ramamurthy V., Scaiano J. C. Principles of molecular photochemistry: an introduction. (University Science Books, 2009).
Lee, D. R. et al. Heavy atom effect of selenium for metal-free phosphorescent light-emitting diodes. Chem. Mater. 32, 2583–2592 (2020).
Article ADS CAS Google Scholar
Zhao, J., Wu, W., Sun, J. & Guo, S. Triplet photosensitizers: from molecular design to applications. Chem. Soc. Rev. 42, 5323–5351 (2013).
Article CAS PubMed Google Scholar
Zhang, X. et al. Recent development of heavy-atom-free triplet photosensitizers: molecular structure design, photophysics and application. J. Mater. Chem. C. 9, 11944–11973 (2021).
Article ADS CAS Google Scholar
Hussain, M. et al. Spin–orbit charge-transfer intersystem crossing of compact naphthalenediimide-carbazole electron-donor–acceptor triads. J. Phys. Chem. B 125, 10813–10831 (2021).
Article CAS PubMed Google Scholar
Dong, Y. et al. Spin–orbit charge-transfer intersystem crossing (SOCT-ISC) in bodipy-phenoxazine dyads: effect of chromophore orientation and conformation restriction on the photophysical
properties. J. Phys. Chem. C. 123, 22793–22811 (2019).
Article CAS Google Scholar
Chen, K. et al. Bodipy derivatives as triplet photosensitizers and the related intersystem crossing mechanisms. Front. Chem. 7, 821 (2019).
Article ADS CAS PubMed Google Scholar
Filatov, M. A. et al. BODIPY-pyrene and perylene dyads as heavy-atom-free singlet oxygen sensitizers. ChemPhotoChem 2, 606–615 (2018).
Article CAS Google Scholar
Buglak, A. A. et al. Quantitative structure-property relationship modelling for the prediction of singlet oxygen generation by heavy-atom-free BODIPY photosensitizers. Chem. Eur. J. 27,
9934–9947 (2021).
Article CAS PubMed Google Scholar
Wang, Z. & Zhao, J. Bodipy–anthracene dyads as triplet photosensitizers: effect of chromophore orientation on triplet-state formation efficiency and application in triplet–triplet
annihilation upconversion. Org. Lett. 19, 4492–4495 (2017).
Article CAS PubMed Google Scholar
Filatov, M. A. et al. Control of triplet state generation in heavy atom-free BODIPY–anthracene dyads by media polarity and structural factors. Phys. Chem. Chem. Phys. 20, 8016–8031 (2018).
Article CAS PubMed Google Scholar
Filatov, M. A. et al. Generation of triplet excited states via photoinduced electron transfer in meso-anthra-BODIPY: fluorogenic response toward singlet oxygen in solution and in vitro. J.
Am. Chem. Soc. 139, 6282–6285 (2017).
Article CAS PubMed Google Scholar
Freeman, D. M. E. et al. Synthesis and exciton dynamics of donor-orthogonal acceptor conjugated polymers: reducing the singlet–triplet energy gap. J. Am. Chem. Soc. 139, 11073–11080 (2017).
Article CAS PubMed Google Scholar
Han, G., Hu, T. & Yi, Y. Reducing the singlet−triplet energy gap by end-group π−π stacking toward high-efficiency organic photovoltaics. Adv. Mater. 32, 2000975 (2020).
Article CAS Google Scholar
Liu, Y., Li, C., Ren, Z., Yan, S. & Bryce, M. R. All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Nat. Rev. Mater. 3, 18020 (2018).
Article ADS CAS Google Scholar
Yu, Y., Mallick, S., Wang, M. & Börjesson, K. Barrier-free reverse-intersystem crossing in organic molecules by strong light-matter coupling. Nat. Commun. 12, 3255 (2021).
Article ADS CAS PubMed PubMed Central Google Scholar
Stranius, K., Hertzog, M. & Börjesson, K. Selective manipulation of electronically excited states through strong light–matter interactions. Nat. Commun. 9, 2273 (2018).
Article ADS PubMed PubMed Central Google Scholar
Cravcenco, A. et al. Exciton delocalization counteracts the energy gap: a new pathway toward NIR-emissive dyes. J. Am. Chem. Soc. 143, 19232–19239 (2021).
Article CAS PubMed PubMed Central Google Scholar
Kautsky, H. & Merkel, H. Phosphoreszenz, Selbstauslöschung und Sensibillisatorwirkung organischer Stoffe. Naturwissenschaften 27, 195–196 (1939).
Article ADS CAS Google Scholar
Kasha, M., Rawls, H. R. & El-Bayoumi, M. A. The exciton model in molecular spectroscopy. Pure Appl. Chem. 11, 371–392 (1965).
Article CAS Google Scholar
Hestand, N. J. & Spano, F. C. Expanded theory of H- and J-molecular aggregates: the effects of vibronic coupling and intermolecular charge transfer. Chem. Rev. 118, 7069–7163 (2018).
Article CAS PubMed Google Scholar
Patalag, L. J., Ho, L. P., Jones, P. G. & Werz, D. B. Ethylene-bridged Oligo-BODIPYs: access to intramolecular J-aggregates and superfluorophores. J. Am. Chem. Soc. 139, 15104–15113 (2017).
Article CAS PubMed Google Scholar
Patalag, L. J., Hoche, J., Mitric, R., Werz, D. B. & Feringa, B. L. Transforming dyes into fluorophores: exciton-induced emission with chain-like Oligo-BODIPY superstructures. Angew. Chem.
Int Ed. 61, e202116834 (2022).
Article ADS CAS Google Scholar
Peng, Y.-Z. et al. Charge transfer from donor to acceptor in conjugated microporous polymer for enhanced photosensitization. Angew. Chem. Int. Ed. 60, 22062–22069 (2021).
Article CAS Google Scholar
Bröring, M. et al. Bis(BF2)-2,2′-Bidipyrrins (BisBODIPYs): highly fluorescent BODIPY dimers with large stokes shifts. Chem. Eur. J. 14, 2976–2983 (2008).
Article PubMed Google Scholar
Cakmak, Y. et al. Designing excited states: theory-guided access to efficient photosensitizers for photodynamic action. Angew. Chem. Int. Ed. 50, 11937–11941 (2011).
Article CAS Google Scholar
Pang, W. et al. Modulating the singlet oxygen generation property of meso–β directly linked BODIPY dimers. Chem. Commun. 48, 5437–5439 (2012).
Article CAS Google Scholar
Hayashi, Y., Yamaguchi, S., Cha, W. Y., Kim, D. & Shinokubo, H. Synthesis of directly connected BODIPY oligomers through Suzuki–Miyaura coupling. Org. Lett. 13, 2992–2995 (2011).
Article CAS PubMed Google Scholar
Nepomnyashchii, A. B., Bröring, M., Ahrens, J. & Bard, A. J. Synthesis, photophysical, electrochemical, and electrogenerated chemiluminescence studies. multiple sequential electron transfers
in BODIPY monomers, dimers, trimers, and polymer. J. Am. Chem. Soc. 133, 8633–8645 (2011).
Article CAS PubMed Google Scholar
Rihn, S., Erdem, M., De Nicola, A., Retailleau, P. & Ziessel, R. Phenyliodine(III) Bis(trifluoroacetate) (PIFA)-promoted synthesis of bodipy dimers displaying unusual redox properties. Org.
Lett. 13, 1916–1919 (2011).
Article CAS PubMed Google Scholar
Lewis, J. & Maroncelli, M. On the (uninteresting) dependence of the absorption and emission transition moments of coumarin 153 on solvent. Chem. Phys. Lett. 282, 197–203 (1998).
Article ADS CAS Google Scholar
Kasha, M. Energy transfer mechanisms and the molecular exciton model for molecular aggregates. Radiat. Res. 20, 55–70 (1963).
Article ADS CAS PubMed Google Scholar
Chai, J.-D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
Article CAS PubMed Google Scholar
Frisch M. J., et al. Gaussian 16 Rev. B.01. (Wallingford, CT; 2016).
Hou, Y. et al. Charge separation, charge recombination, long-lived charge transfer state formation and intersystem crossing in organic electron donor/acceptor dyads. J. Mater. Chem. C. 7,
12048–12074 (2019).
Article CAS Google Scholar
Wang, Z. et al. Insight into the drastically different triplet lifetimes of BODIPY obtained by optical/magnetic spectroscopy and theoretical computations. Chem. Sci. 12, 2829–2840 (2021).
Article CAS Google Scholar
Englman, R. & Jortner, J. The energy gap law for non-radiative decay in large molecules. J. Lumin 1-2, 134–142 (1970).
Article Google Scholar
Englman, R. & Jortner, J. The energy gap law for radiationless transitions in large molecules. Mol. Phys. 18, 145–164 (1970).
Article ADS CAS Google Scholar
Siebrand, W. Radiationless transitions in polyatomic molecules. II. triplet‐ground‐state transitions in aromatic hydrocarbons. J. Chem. Phys. 47, 2411–2422 (2004).
Article ADS Google Scholar
Scharf, B. & Dinur, U. Striking dependence of the rate of electronic radiationless transitions on the size of the molecular system. Chem. Phys. Lett. 105, 78–82 (1984).
Article ADS CAS Google Scholar
Humeniuk, A., Mitrić, R. & Bonačić-Koutecký, V. Size dependence of non-radiative decay rates in J-aggregates. J. Phys. Chem. A 124, 10143–10151 (2020).
Article CAS PubMed Google Scholar
Poriel, C. et al. Dispirofluorene–indenofluorene derivatives as new building blocks for blue organic electroluminescent devices and electroactive polymers. Chem. Eur. J. 13, 10055–10069
(2007).
Article CAS PubMed Google Scholar
Brouwer, A. M. Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report). Pure Appl. Chem. 83, 2213–2228 (2011).
Article CAS Google Scholar
Hankache, J., Niemi, M., Lemmetyinen, H. & Wenger, O. S. Hydrogen-bonding effects on the formation and lifetimes of charge-separated states in molecular triads. J. Phys. Chem. A 116,
8159–8168 (2012).
Article CAS PubMed Google Scholar
Haefele, A., Blumhoff, J., Khnayzer, R. S. & Castellano, F. N. Getting to the (square) root of the problem: how to make noncoherent pumped upconversion linear. J. Phys. Chem. Lett. 3,
299–303 (2012).
Article CAS Google Scholar
Download references
AcknowledgementsThe Swedish NMR center is acknowledged for access to high-field NMRs. The authors especially acknowledge Zoltan Takacs for performing diffusion NMR spectroscopy and analysis thereof. K.B.
gratefully acknowledge financial support from the Knut and Alice Wallenberg Foundation (KAW 2017,0192). B.A. acknowledges the Swedish research council (VR) for support. Our research relied
on computational resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at C3SE and NSC partially funded by the Swedish research council through
grant agreement no. 2022-06725.
FundingOpen access funding provided by University of Gothenburg.
Author informationAuthor notesThese authors contributed equally: Clara Schäfer, Rasmus Ringström.
Authors and Affiliations Department of Chemistry and Molecular Biology, University of Gothenburg, Box 462, 405 30, Gothenburg, Sweden
Clara Schäfer & Karl Börjesson
Department of Chemistry and Chemical Engineering, Chalmers University of Technology, Kemivägen 10, 412 96, Gothenburg, Sweden
Rasmus Ringström, Martin Rahm & Bo Albinsson
Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, Sahlgrenska Academy at the University of Gothenburg, Mölndal Hospital, House V3, 431 80, Mölndal,
Sweden
Jörg Hanrieder
Department of Neurodegenerative Disease, Queen Square Institute of Neurology, University College London, London, WC1N 3BG, London, UK
Jörg Hanrieder
AuthorsClara SchäferView author publications You can also search for this author inPubMed Google Scholar
Rasmus RingströmView author publications You can also search for this author inPubMed Google Scholar
Jörg HanriederView author publications You can also search for this author inPubMed Google Scholar
Martin RahmView author publications You can also search for this author inPubMed Google Scholar
Bo AlbinssonView author publications You can also search for this author inPubMed Google Scholar
Karl BörjessonView author publications You can also search for this author inPubMed Google Scholar
ContributionsK.B. together with C.S. designed the project idea and the molecules. C.S. synthesized and analyzed the molecules, and performed absorption, emission and initial transient absorption
experiments and analysis thereof. R.R. performed electrochemical analysis of the compounds as well as transient absorption measurements. M.R. performed DFT calculations. B.A., K.B., R.R.,
and C.S. analyzed all collected results. J.H. performed MALDI analysis of the synthesized BODIPY oligomers. All authors contributed to writing the manuscript. All authors have given approval
to the final version of the manuscript.
Corresponding author Correspondence to Karl Börjesson.
Ethics declarations Competing interestsThe authors declare no competing interest.
Peer review Peer review informationNature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional informationPublisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary informationSupplementaryInformationPeer Review FileDescription of Additional Supplementary FilesSupplementary Dataset 1Supplementary Dataset 2Source dataSource DataRights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or
other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and permissions
About this articleCite this article Schäfer, C., Ringström, R., Hanrieder, J. et al. Lowering of the singlet-triplet energy gap via intramolecular exciton-exciton coupling. Nat Commun 15,
8705 (2024). https://doi.org/10.1038/s41467-024-53122-7
Download citation
Received: 24 November 2023
Accepted: 27 September 2024
Published: 08 October 2024
DOI: https://doi.org/10.1038/s41467-024-53122-7
Share this article Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article.
Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
The government's net zero emissions target comes back to bite | thearticleA court has ruled that the planned expansion of Heathrow Airport is unlawful because it does not take climate commitment...
Redistributing Foreign Aid - Los Angeles TimesIt was colonels like Summers with their paranoia of communism who advised U.S. leaders to squander untold billions on us...
Michael Schumacher: How is the Legendary Racer's Health Now?One of the most loved personalities in the sporting world, Michael Schumacher, turns 53 on Monday, 3 January. The seven-...
Canary Wharf station CLOSED due to power failure - SEVERE delays on Jubilee LineTransport for London has confirmed that services between Finchley Road and Stratford are experiencing long delays follow...
8 Saved Near Burning Boat - Los Angeles TimesL.A. Times Archives June 13, 1990 12 AM PT Share via Close extra sharing options Email Facebook X LinkedIn Threads Reddi...
Latests News
Lowering of the singlet-triplet energy gap via intramolecular exciton-exciton couplingDownload PDF Article Open access Published: 08 October 2024 Lowering of the singlet-triplet energy gap via intramolecula...
The Nicotine Network (cont’d)Ramhurst grew out of an earlier operation, set up by RJR executive Mike Phillips in the mid-1980s. Phillips’ team hosted...
florim's 'heritage luxe' collection exuberates european palace marble divinityThe divinity of European palaces dressed in marble takes a modern twist as Florim introduces Heritage Luxe. Using the co...
Mark-paul gosselaar: from ‘saved by the bell’ nice guy to kidnapper on ‘found’ | members onlyMark-Paul Gosselaar, 50, has been working steadily since he rose to fame 30-some years ago playing Zack Morris on _Save...
If not mother then who’ll you salute, afzal guru? Asks venkaiahWhen someone says 'Bharat Mata ki jai' it is not only about some goddess in a photo, Naidu said. "It is a...