Coronavirus envelope protein activates tmed10-mediated unconventional secretion of inflammatory factors
Coronavirus envelope protein activates tmed10-mediated unconventional secretion of inflammatory factors"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The precise cellular mechanisms underlying heightened proinflammatory cytokine production during coronavirus infection remain incompletely understood. Here we identify the envelope
(E) protein in severe coronaviruses (SARS-CoV-2, SARS, or MERS) as a potent inducer of interleukin-1 release, intensifying lung inflammation through the activation of TMED10-mediated
unconventional protein secretion (UcPS). In contrast, the E protein of mild coronaviruses (229E, HKU1, or OC43) demonstrates a less pronounced effect. The E protein of severe coronaviruses
contains an SS/DS motif, which is not present in milder strains and facilitates interaction with TMED10. This interaction enhances TMED10-oligomerization, facilitating UcPS cargo
translocation into the ER-Golgi intermediate compartment (ERGIC)—a pivotal step in interleukin-1 UcPS. Progesterone analogues were identified as compounds inhibiting E-enhanced release of
proinflammatory factors and lung inflammation in a Mouse Hepatitis Virus (MHV) infection model. These findings elucidate a molecular mechanism driving coronavirus-induced hyperinflammation,
proposing the E-TMED10 interaction as a potential therapeutic target to counteract the adverse effects of coronavirus-induced inflammation. SIMILAR CONTENT BEING VIEWED BY OTHERS
IMMUNOLOGICAL MECHANISMS OF THE NUCLEOCAPSID PROTEIN IN COVID-19 Article Open access 14 February 2024 THE WNT/Β-CATENIN PATHWAY IS IMPORTANT FOR REPLICATION OF SARS-COV-2 AND OTHER
PATHOGENIC RNA VIRUSES Article Open access 21 February 2024 CORONAVIRUSES EXPLOIT A HOST CYSTEINE-ASPARTIC PROTEASE FOR REPLICATION Article 03 August 2022 INTRODUCTION Excessive and
uncontrolled proinflammatory cytokine production, notably evident in pathogen infections such as coronavirus (e.g., SARS-CoV-2, SARS, or MERS), can precipitate immunopathogenesis, resulting
in widespread tissue damage, including acute lung injuries, across the human body1,2,3,4,5,6. This process correlates with the emergence of multiple organ dysfunction syndrome and an
elevated likelihood of mortality1,2,3,4,5. The observed surge in proinflammatory cytokines, including interleukin-1 (IL1), interleukin-6 (IL6), interleukin-12 (IL12), interferon-gamma
(IFNγ), and tumor necrosis factor-alpha (TNFα) circulates in the bloodstream. While these cytokines initially function as protective signals to activate the immune system, their excessive
release significantly exacerbates the severity of the condition2,3,6,7,8,9. Among these inflammatory mediators, members of the IL1 family, notably IL1β and IL33, serve as crucial upstream
regulators in the inflammatory cascade10,11,12. Prior studies on protein-protein interactions have indicated the potential for viral proteins from coronaviruses (e.g., SARS-CoV-2) to provoke
the release of IL1s, including IL1β and IL3313,14,15, both recognized for their engagement in unconventional protein secretion16,17,18. Gasdermins have been identified as key regulators in
the release of IL1β and IL33. For example, acute bacterial infection leads to Gasdermin D (GSDMD) -dependent release of IL1β in macrophages, typically accompanied by pyroptosis19,20,21.
Allergens can trigger GSDMD-mediated release of IL33 in epithelial cells22, and helminth infection results in GSDMC-dependent release of IL33 in the intestin23. Additionally, the release of
IL1β and IL33 can be regulated by GSDMD-independent pathways, including TMED10-mediated unconventional secretion, secretory autophagy and PIP2-dependent translocation like FGF217,24,25,26.
Nevertheless, a comprehensive molecular elucidation regarding how coronaviruses induce the unconventional release of inflammatory factors remains elusive, particularly considering the
variability in infection outcomes among distinct coronaviruses. Lethal strains such as SARS-CoV-2, SARS-CoV, or MERS-CoV prompt heightened proinflammatory cytokine production, whereas
coronaviruses like 229E, HKU1, or OC43 result in mild symptoms with reduced inflammation27,28,29. The intricate cellular mechanisms governing coronavirus modulation of host inflammatory
responses, particularly the release of proinflammatory factors, remain incompletely understood, including the interplay between viral factors and host secretion systems leading to excessive
inflammatory factor release at cellular and subcellular levels, and the distinctions between severe and mild viruses in this regard. An intriguing aspect of coronavirus biology involves the
utilization of the ER-Golgi intermediate compartment (ERGIC) as a packaging site, where various viral factors accumulate30,31,32. Previously, we identified a protein translocation pathway
operating within the ERGIC, responsible for the unconventional release of multiple inflammatory factors, including members of the IL1 family17. This pathway, termed TMED10-channeled
unconventional protein secretion (THU), relies on the ERGIC-localized TMED10 protein to facilitate the entry of IL1 family factors into the secretory system, initiating unconventional
secretion17. Our prior investigations underscored the regulatory role of THU in inflammatory responses to bacterial infections and colitis17,33. In this study, we discovered that the E
proteins from severe coronaviruses (SARS-CoV-2, SARS, and MERS) markedly stimulate the release of inflammatory factors, exacerbating lung inflammation through THU, whereas mild strains exert
less impact. Mechanistically, the interaction between E proteins and TMED10 promotes TMED10 oligomerization, facilitating the translocation of UcPS cargo into the ERGIC—an essential step in
interleukin-1 UcPS. We identified a specific SS/DS motif on the E proteins of severe coronaviruses crucial for TMED10 interaction and the subsequent triggering of excessive inflammatory
factor release. Significantly, through screening of FDA-approved drugs, we identified progesterone analogs that mainly function to disrupt the E-TMED10 interaction, effectively reducing
proinflammatory factor release and lung inflammation in a MHV infection model. These findings highlight the E-TMED10 interaction as a promising therapeutic target, with progesterone analogs
showing potential as lead compounds for developing anti-hyperinflammatory agents to mitigate the adverse effects of coronavirus-induced inflammation. RESULTS CORONAVIRUS E PROTEIN PROMOTES
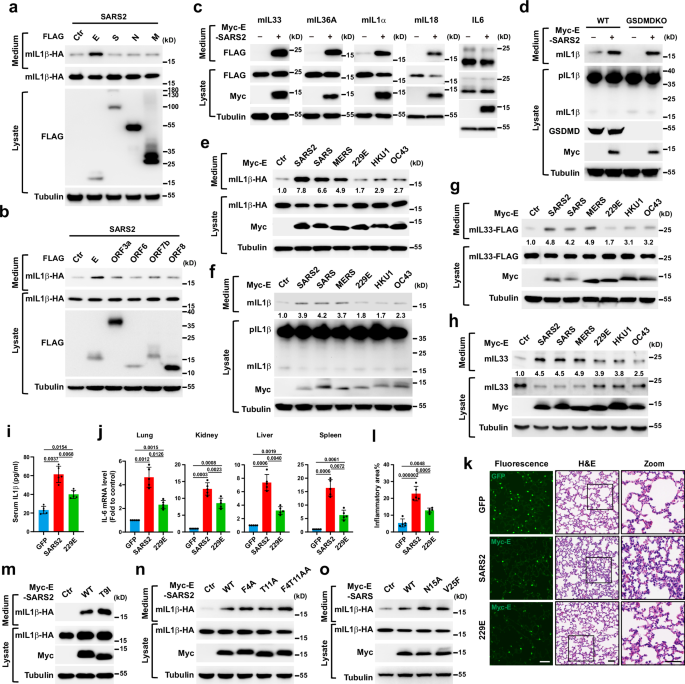
UCPS To elucidate the mechanisms by which coronavirus proteins induce UcPS of inflammatory factors, we conducted an examination focusing on the influence of individual SARS2 proteins on the
secretion of mature IL1β (mIL1β). This assessment employed a previously established secretion system34.Remarkably, the expression of the E protein was found to enhance IL1β secretion, while
the other SARS-CoV-2 factors (N, S, M, ORF3a, ORF6, ORF7b, and ORF8) exhibited comparatively lesser effects (Fig. 1a, b). The effect of the E protein on IL1β secretion was found to be
dose-dependent (Supplementary Fig. 1a), and this effect was independent of the specific tag used, as both untagged and Myc or Flag-tagged E consistently enhanced IL1β secretion
(Supplementary Fig. 1b). Additionally, the E protein also promoted the UcPS of other IL1 family inflammatory factors, including IL33, IL36α, IL1α, and IL18, while it had no influence on the
secretion of conventional cargo IL6 (Fig. 1c). Previous studies showed SARS-CoV-2 infection can cause host inflammation through GSDMD-mediated pyroptosis by activating inflammasomes, which
cleave GSDMD, triggering cell lysis and the release of pro-inflammatory cytokines including IL1β and IL18, thereby amplifying the inflammatory response35,36,37. Interestingly, when E protein
expression was tested in GSDMD knockout THP-1 cells, it was found to enhance the secretion of mature IL1β, suggesting that E-regulated UcPS is independent of GSDMD (Fig. 1d). Furthermore, E
protein expression had minimal effect on Caspase-3 cleavage or LDH release, indicating that the E-regulated UcPS is not attributed to cell death (Supplementary Fig. 1a). To investigate
whether E proteins from various coronaviruses impact UcPS, we assessed the secretion of mIL1β in both non-inflammatory and inflammatory cell contexts. Surprisingly, we observed that E
proteins from severe symptom-causing coronaviruses (Es-SSC), including SARS-CoV-2, SARS, and MERS, strongly enhanced mIL1β secretion, while those from mild symptom-causing coronaviruses
(Es-MSC), such as 229E, HKU1, and OC43, had a moderate effect (Fig. 1e, f). A similar trend was noted in the stimulation of mIL33 secretion, both in non-inflammatory cells and BEAS-2B lung
bronchial epithelial cells (Fig. 1g, h). To elucidate the role of the E protein in promoting inflammation within physiological settings, we expressed the E proteins of SARS-CoV-2 (E-SARS2,
an example of Es-SSC) and 229E (E-229E, an example of Es-MSC) in the lungs of C57BL/6 mice using Adeno-Associated Virus (AAV)38 (Fig. 1k, left panel). Following a moderate challenge with
lipopolysaccharide (LPS), we found that E-SARS2, but not E-229E, expression led to an increase in inflammation, evidenced by elevated serum levels of IL1β (Fig. 1i). This was accompanied by
a corresponding rise in IL6 production in the lung, kidney, liver, and spleen (Fig. 1j). Additionally, the expression of E-SARS2, as opposed to E-229E, enhanced the infiltration of immune
cells into the lung (Fig. 1k, middle and right panel, and 1 l). These findings suggest that coronavirus E proteins promote the UcPS of inflammatory factors, and the impact of different E
proteins correlates with the severity of symptoms following infection. Previously documented evidence has established the coronavirus E protein’s role as a viroporin5,39,40,41,42,43, forming
an ion channel capable of activating the inflammasome and potentially enhancing the maturation and secretion of IL1β44,45. Contrary to expectations, both E-SARS2 and E-SARS, even when
engineered with mutations eliminating ion channel activity5,41,46,47,48, exhibited the same efficiency in promoting the secretion of mIL1β-HA as the wild-type (WT) E protein in
non-inflammatory cells (Fig. 1m–o). These findings suggest that ion channel activity is unlikely to directly regulate the secretion of mIL1β itself. Notably, the T9I mutation in the E
protein, present in the omicron strain of SARS-CoV-242,49, did not impact the effect of E on UcPS (Fig. 1m). However, this mutation significantly decreased stability (Supplementary Fig. 1c,
d), potentially compromising sustained UcPS triggered by E. The destabilization together with a compromised channel activity likely contributes to decreased inflammation upon omicron
infection42,50,51. E-REGULATED UCPS IS DEPENDENT ON TMED10 Previous research has established that the coronavirus E protein primarily localizes to the ERGIC31,32,43,52, a compartment we’ve
identified as a pivotal station in the regulation of IL1 family members’ secretion through an UcPS pathway termed TMED10-channeled UcPS (THU)17. Importantly, E proteins from both severe and
mild symptom-causing coronaviruses are found to be concentrated on the ERGIC (Fig. 2a). Notably, E-SARS2 was observed to co-localize with TMED10 on the ERGIC (Fig. 2b), suggesting that
E-mediated regulation of UcPS may be linked to the THU pathway. Intriguingly, E-SARS2 failed to enhance the secretion of mIL1β in TMED10 knockout (KO) cells (Fig. 2c), a pattern similarly
observed in the secretion of mIL33 and mIL36α (Supplementary Fig. 2a, b). Further analysis of endogenous mIL1β and mIL33 secretion in THP-1 or BEAS-2B cells revealed that E-regulated UcPS
was substantially impaired in the absence of TMED10, an effect that could be restored upon TMED10 re-expression (Fig. 2d, e). This finding further confirms that E-SARS2 promotes UcPS through
the THU pathway. In mice lacking TMED10, the expression of E-SARS2 failed to increase serum IL1β levels, as well as the production of IL6 and lung injury resulting from immune cell
infiltration, in comparison to their WT littermates (Fig. 2f–i). Collectively, these results provide strong evidence that E-SARS2 promotes the UcPS of mIL1β and other inflammatory factors
through the THU pathway. We sought to understand how the coronavirus E protein regulates UcPS through the THU. Our investigations revealed that the E-SARS2 protein exhibited a notable
association with TMED10, as observed in co-immunoprecipitation (Co-IP) and GST-pull down experiments (Fig. 2j, k). Notably, E-SARS2 showed a reduced interaction with TMED10 lacking the
C-terminal domain (TM10ΔCT mutant) (Fig. 2j). Furthermore, in a pull-down experiment, E-SARS2 was found to specifically interact with the C-terminal (CT) peptide of TMED10, but not with the
CT of TMED6 (Fig. 2l). These findings strongly suggest a direct binding interaction between TMED10-CT and E-SARS2. In TMED10 knockout (TMED10KO) THP-1 cells, E-SARS2 effectively increased
the secretion of IL1β when full-length TMED10 was reintroduced, but this rescue was not achieved when the TM10ΔCT variant was utilized (Fig. 2m). These findings underscore the indispensable
role of the association between TMED10’s C-terminal domain (TMED10-CT) and E-SARS2 in the regulation of E-SARS2-mediated UcPS. Previous research from our group had indicated that TMED10 has
the capacity to form oligomers in the presence of secretory cargo, potentially serving as a protein channel-like machinery responsible for the regulation of protein translocation17.
Interestingly, E-SARS2 expression facilitates the self-association of TMED10 and boosts the formation of TMED10 oligomers in the presence and absence of IL1β, without affecting its
interaction with another TMED family member, TMED2 (Supplementary Fig. 2c, d). This effect was contingent on the interaction between TMED10 and E-SARS2, as the oligomerization of the TM10ΔCT
variant remained unaffected by E-SARS2 expression (Supplementary Fig. 2e). By using crosslinking, we were able to detect a ~ 38KDa band of E in the presence of TMED10, indicating to be a
heterodimer of E and TMED10 (Supplementary Fig. 2f). However, we could barely detect the presence of E in high molecular weight bands (Supplementary Fig. 2f). The data suggest that E binds
TMED10 which may make TMED10 monomer prone to form oligomers likely via enhancing cargo TMED10 binding as well as other unknown reasons that trigger conformation change of TMED10 favoring
oligomerization. We also observed that the binding of the E protein with TMED10 does not inhibit but rather enhances the association between TMED10 and IL1β (Supplementary Fig. 2g). This
could be due to the E protein promoting the formation of TMED10 homo-oligomers, which increases the number of cargo binding sites through multiple C-terminal tails in the oligomer.
Crucially, Es-SSC exhibited a stronger association with TMED10 compared to Es-MSC (Fig. 2n), and consequently more effectively increased TMED10 oligomerization (Fig. 2o). These results
suggest that TMED10 may represent a host target for Es-SSC in the facilitation of UcPS for inflammatory factors. E FACILITATES CARGO MEMBRANE TRANSLOCATION In THU, TMED10 serves the
essential role of facilitating the translocation of cargoes into the ERGIC17. This process involves ushering leaderless UcPS cargo into the vesicle trafficking system to initiate the UcPS
pathway within the cell. To assess whether the E protein supports the entry of secretory cargo into vesicles, an established in vitro membrane translocation assay was conducted (Fig. 3a). As
previously demonstrated, the presence of TMED10 on the liposome effectively promoted the translocation of mIL1β into the liposome, as indicated by its protection from proteinase K
digestion. Notably, E-SARS2 alone in the liposome exhibited limited cargo translocation ability, but in combination with TMED10, it significantly augmented TMED10-facilitated translocation
(Fig. 3b, Supplementary Fig. 3a). A comparable enhancement of TMED10-mediated translocation was also observed with E-SARS (Supplementary Fig. 3a, b). These findings substantiate that both
E-SARS2 and E-SARS directly enhance the translocation of UcPS cargo into vesicles via TMED10. Given the unavailability of E protein from other coronaviruses, a cell-free UcPS cargo
translocation assay was developed utilizing membrane fractions from cells expressing various E proteins (Fig. 3c). Similar to the liposome assay, E-SARS2 elevated mIL1β translocation in the
presence of TMED10 (Fig. 3d). Additionally, Es-SSC promoted mIL1β translocation, whereas Es-MSC exhibited a lesser impact on mIL1β translocation (Fig. 3e). To further corroborate the
differential effects of E proteins on cargo entry into vesicles, a GFP complementation assay was employed, as previously established17, to assess UcPS cargo translocation into the ERGIC
(Fig. 3f). In this assay, a GFP1-10 fragment was fused to the luminal segment of TMED10 to evaluate the translocation of GFP11-tagged mIL1β into the ERGIC lumen, where GFP complementation
occurs. Once again, Es-SSC, rather than Es-MSC, was found to enhance mIL1β entry into the ERGIC (Fig. 3g, Supplementary Fig. 3c). These results collectively suggest that Es-SSC augments the
UcPS of inflammatory factors by promoting cargo translocation within THU. AN SS/DS MOTIF IN ES-SSC DETERMINES THEIR EFFECT ON TMED10-MEDIATED UCPS Sequence alignment revealed the presence of
an SS/DS motif in Es-SSC but its absence in Es-MSC (Fig. 4a). The alteration of the SS/DS motif minimally impacted the ERGIC localization of Es-SSC (Supplementary Fig. 4a). However, this
mutation significantly compromised Es-SSC’s capacity to enhance mIL1β secretion in both non-inflammatory and inflammatory cells (Fig. 4b, c), hindered its association with TMED10, and its
ability to facilitate cargo translocation (E-SARS2) (Fig. 4d, e). This suggests that the SS/DS motif is essential for Es-SSC to augment the THU pathway. Conversely, the introduction of the
SS motif into Es-MSC increased their interaction with TMED10, regulated secretion, and enhanced cargo translocation (Fig. 4f–k). It is conceivable that pathogenic coronavirus E proteins may
possess an SS/DS motif that plays a crucial role in promoting UcPS of inflammatory factors. E-TMED10 INTERACTION IS REQUIRED FOR E-INDUCED INFLAMMATION The presented data suggests that the
interaction between E and TMED10 initiates TMED10 oligomerization and cargo translocation, resulting in the release of various inflammatory factors through the THU pathway. Our objective was
to investigate whether blocking the E-TMED10 complex could mitigate inflammation induced by E expression. E is a small protein characterized by a luminal N-terminal domain, a transmembrane
domain, and a C-terminal (CT) cytosolic domain (Fig. 5a)31. The association of E-SARS2 with TMED10-CT suggests a potential interaction between the cytosolic portion of E-SARS2 and the CT of
TMED10. To disrupt the E-TMED10 interaction, we overexpressed TMED10-CT or E-SARS2-CT. Remarkably, the expression of green fluorescent protein-tagged E-SARS2-CT (GFP-ECT), as opposed to
TMED10-CT expression, effectively impeded the E-TMED10 interaction (Fig. 5b, Supplementary Fig. 4b). Consistently, the expression of GFP-ECT demonstrated a dose-dependent inhibition of
E-SARS2 induced mIL1β secretion in both HEK293T cells and THP-1 cells (Fig. 5c, d). In experiments involving crosslinking and translocation, GFP-ECT reduced E-facilitated TMED10
oligomerization and the translocation of mIL1β into the membrane fraction (Fig. 5e, f). In a mouse model, the expression of GFP-ECT resulted in diminished inflammation, as indicated by
reduced levels of IL1β in the serum and correspondingly reduced production of IL6 in the lung, kidney, liver, and spleen (Fig. 5g–j). These findings emphasize the essential role of E-TMED10
interaction in facilitating E-SARS2-induced release of inflammatory factors through the THU pathway. PROGESTERONE AND ITS ANALOGS INHIBIT E-INDUCED INFLAMMATORY FACTOR RELEASE Our findings
suggest that the E protein’s induction of inflammatory factor release via the THU pathway may contribute to the development of severe inflammation in SARS-CoV-2 and other coronaviruses,
leading to severe symptoms. Subsequently, we conducted a search for inhibitors against E-induced UcPS, which could serve as a potential target for mitigating severe inflammation resulting
from coronavirus infections. To facilitate this investigation, we established a rapid secretion analysis assay using complementary NanoLuc luciferase53. This assay was then combined with
high-throughput compound screening to identify chemical modulators of E-mediated UcPS (Fig. 6a). Out of approximately 2500 FDA-approved drugs, we discovered that progesterone and some of its
analogs exhibited strong inhibitory effects on E-stimulated UcPS (Fig. 6b, c). Both progesterone and its analog Ulipristal acetate (UPA) effectively suppressed the release of IL1β induced
by E-SARS2-SSC in THP-1 cells (Fig. 6d, e). Similar to the ECT-peptide, progesterone and UPA blocked the interaction between E and TMED10, inhibited E-induced TMED10 oligomer formation, and
prevented E-enhanced mIL1β translocation (Fig. 6f–h). These findings suggest that these two drugs may function through a mechanism similar to ECT. In a mouse experiment, UPA reduced lung
inflammation caused by the expression of E-SARS2 (Fig. 6i–l). Consequently, we conclude that progesterone and its analogs inhibit E-induced UcPS by disrupting the interaction between E and
TMED10. To further elucidate the structural features necessary for the inhibitory function of progesterone and its analogs in regulating UcPS mediated by TMED10, we evaluated the effects of
23 progesterone analogs on E-enhanced mIL1β secretion (Fig. 6m, Supplementary Data 1). Interestingly, we observed that modifications at position 11 on the steroid core of progesterone
analogs, such as the introduction of a carbonyl or hydroxyl group, largely abolished their inhibitory effects (Fig. 6m). However, the presence of an N, N-dimethylaniline modification at
position 11, as seen in UPA, did not diminish the inhibitory effect (Fig. 6m). Furthermore, we observed various modifications at position 17 within the progesterone analogs, but these
modifications did not significantly impact the activity of these compounds in regulating E-enhanced UcPS in the secretion assay (Fig. 6n). TMED10 CONTROLS MHV-INDUCED UCPS OF INFLAMMATORY
FACTORS In order to investigate the role of TMED10-regulated UcPS in the release of inflammatory factors during coronavirus infection, we established an infection model using Murine
Hepatitis Virus (MHV) in HEK293T cells and bone marrow-derived macrophages (BMDMs)54. Similar to the effects observed with E protein expression, MHV infection elicited a dose-dependent
release of mIL1β in HEK293T cells expressing the MHV receptor mCC1a (Fig. 7a). Importantly, we observed that Caspase-3 cleavage, indicative of cell death, did not increase during MHV
infection, implying that mIL1β release was more likely attributed to UcPS rather than cell death (Fig. 7a). In the case of BMDMs, the release of mIL1β induced by MHV infection was found to
be dependent on TMED10 rather than GSDMD (Fig. 7b, c), underscoring the involvement of the THU pathway. Furthermore, the expression of ECT or the administration of progesterone (or UPA)
effectively blocked the MHV infection-induced mIL1β secretion and the interaction between E-MHV and TMED10 (Fig. 7d–g), highlighting the significance of the E-TMED10 interaction in this
process. In a murine lung infection model using IFNAR-KO mice55,56, MHV infection resulted in severe lung infection, characterized by the release of mIL1β, immune cell infiltration, and the
activation of IL6 production in multiple organs (Fig. 7h–o). These effects mirrored those seen with E protein expression in the presence of mild LPS challenges. Significantly, the expression
of ECT or treatment with progesterone (or UPA) mitigated the inflammation following MHV infection, further substantiating the role of these interventions in modulating the inflammatory
response (Fig. 7h–o). The mitigating effect of progesterone on inflammation induced by coronavirus was reported in two other studies, aligning with our current findings57,58. DISCUSSION In
summary, our research uncovers a crucial molecular interaction, namely the E-TMED10 interaction, which triggers the THU-mediated release of inflammatory factors and consequently results in
severe inflammation during coronavirus infections. Our findings from cellular, molecular, and murine experiments, strongly underscores the potential of targeting the E-TMED10 interaction as
a therapeutic strategy to combat the lethal cytokine storm induced by SARS-CoV-2 and other highly virulent coronaviruses. The coronavirus E protein has garnered attention as a key structural
component implicated in various stages of the virus lifecycle and directly linked to pathogenic processes59,60. Multiple investigations have highlighted its ability to form ion channels,
leading to cell death, particularly noteworthy in inflammatory cells where E-induced ion channels activate the inflammasome, prompting pyroptosis5,44,45,46. Furthermore, surface-localized E
proteins of SARS-CoV-2 can activate TLR2, eliciting an inflammatory response61. Additionally, E proteins can interact with cell junction proteins, disrupting epithelial cell junctions and
causing tissue leakage and damage62,63. Consistent with these findings, our research supports the concept that the E protein serves as a pathogenic factor contributing to tissue damage and
inflammation. Furthermore, our investigation sheds light on the differential host inflammatory responses triggered by severe and mild coronaviruses. At the molecular level, we propose a
model in which E proteins from severe-symptom-causing coronaviruses, including SARS-CoV-2, SARS and MERS, interact with TMED10 to activate THU-mediated release of inflammatory cytokines
(Fig. 7p). Mechanistically, a specific SS/DS motif present in severe coronaviruses, absent in mild counterparts, enhances interaction with TMED10, promoting its oligomerization—a critical
step in the translocation process essential for the UcPS of various inflammatory factors. Furthermore, it is noteworthy that coronavirus E proteins can form ion channels, capable of
triggering inflammasome activation5,44,45. In this context, the pro-inflammatory role of E proteins aligns with their facilitation of inflammatory factor release, collectively contributing
to the extensive release of such factors as a risk factor for the development of severe symptoms in coronavirus infections. Recent studies have demonstrated that SARS-CoV-2 ORF8, a protein
equipped with a signal peptide, can be secreted through both conventional and unconventional pathways64,65. Notably, the unconventional secretion pathway allows ORF8 to evade glycosylation,
resulting in the release of an unglycosylated form of the protein64. This unglycosylated ORF8 has been implicated in the induction of pro-inflammatory cytokines by binding to the IL17RA
receptor64,66. Investigating the regulatory mechanisms that govern these distinct ORF8 secretion pathways is of significant scientific interest. It remains to be elucidated whether the
unconventional secretion of ORF8 is regulated through TMED10-mediated translocation. A characteristic of viral infection at the cellular level involves the manipulation of the host cellular
machinery, leading to disruptions in cellular function that can potentially be targeted for antiviral interventions. In the context of SARS-CoV-2 infection, the virus exploits the host
autophagy-lysosome system for release through ORF3a, repurposing degradative autophagy for secretion54,67. Additionally, Nsp3 and Nsp4 collaborate with host factors VMP1 and TMEM41B to
modify the endoplasmic reticulum (ER) membrane, forming double-membrane vesicles crucial for virus replication68,69,70. Our research adds to this understanding by demonstrating that the E
protein of various coronaviruses interacts with TMED10, triggering host inflammation, and suggests that disrupting the E-TMED10 interaction could be a viable anti-inflammatory strategy.
Notably, we discovered that progesterone analogs inhibit the E-TMED10 interaction, offering protection against hyperinflammatory damage induced by both E protein expression and coronavirus
infection. This finding provides insight into the previously unexplained gender differences observed in COVID-19 severity, where males exhibit higher severity and fatality rates compared to
females71. Given that the E-TMED10 interaction represents a common target for inflammation modulation among severe coronaviruses, progesterone analogs may serve as promising lead compounds
for the development of novel anti-inflammatory drugs against current COVID 19 and future pandemics caused by new deadly coronaviruses. It is noteworthy that while our findings suggest the
inhibition of the E-TMED10 interaction as a potential target for progesterone analogs, it remains uncertain whether other unidentified targets exist in the case of proinflammatory factor
UcPS. Our study on how viral factors influence the release of non-classical inflammatory cytokines has opened up new avenues of investigation into how pathogen-host interactions lead to
inflammation, which can guide the study of inflammation caused by other viruses, such as influenza, HBV, and Zika. METHODS This study complied with all of the relevant ethical regulations.
All mice experiments were approved by the Institutional Animal Care and Use Committees at Tsinghua University (permission number: 22-ZM5). PLASMIDS AND CELLS The mature form of IL1 family
proteins (IL1α, IL1β, IL18, IL33, IL36α), IL6, TMED10-V5, TMED10∆CT-V5, HA-TMED10, GFP (1-10)-TMED10-V5, mIL1β-FLAG-GFP11 plasmids and mIL1β, TMED10 protein purification plasmids were
generated in our previous work17. FLAG-tagged expression plasmids of SARS2 proteins (E, M, S, N, ORF3a, ORF6, ORF7b, and ORF8) were described as previously72. E-SARS2 expression plasmids
with or without an N-terminal Myc tag were PCR amplified from the template and inserted into the FUGW vector. Expression plasmids of E proteins of SARS, MERS, 229E, HKU1, OC43 and MHV were
generated by DNA synthesize followed by inserting into the FUGW vector with a Myc tag at the N terminus. Mutants of E proteins were constructed by mutagenesis PCR. GFP-ECT was generated by
PCR amplification from 38-71aa of E-SARS2 C-terminal and inserted into the FUGW vector with a GFP tag at the N terminus. Myc-E-SARS2, Myc-E-SARS were also inserted into pGEX4T1 or pET28a
vector with a GST or MBP tag for protein purification. HEK293T (from Dr. Randy Schekman), TMED10-KO HEK293T (from our laboratory), U2OS (from Dr. Randy Schekman), BEAS-2B (from Dr. Yu Rao)
and 17Cl-1 (from Dr. Fuping You) cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and 1% Pen-Strep. THP1 (from Dr. Gong Cheng),
TMED10-KO (from our laboratory) and GSDMD-KO THP1 (from our laboratory) cells were maintained in RPMI-1640 medium supplemented with 10% FBS and 1% Pen-Strep. The cells were cultured at 37 °C
with 5% CO2. Bone marrow-derived macrophages (BMDMs) were isolated from 6-week-old male C57BL/6 mice and differentiated using standard protocols73. REAGENTS AND ANTIBODIES The reagents used
in this study were purchased from the following sources: DSS (Thermo, 21655), GTP (Roche, 11140957001), ATP (Sigma, A2383), Creatine phosphate (Calbiochem, 2380), Creatine Kinase (Roche,
10127566001), Proteinase K (ABCone, P78893), Phenylmethylsulfonyl fluoride (PMSF) (Beyotime, ST505), Opti-Prep (Serumwerk Bernburg AG, 1893), Protease inhibitor cocktail (Roche,
11697498001), Phosphotase inhibitors (Roche, 4906845001), LPS (Sigma, L2880), Mouse IL-1 beta Uncoated ELISA Kit (Thermo, 88-7013-22), anti-V5 agarose (Sigma, A7345), anti-Myc agarose
(Thermo, 20168). Progesterone (S1705), Nitazoxanide (S1627), Leflunomide (S1247), Nitrendipine (S2491), Lomerizine 2HCl (S4084), Econazole Nitrate (S2535), Hexachlorophene (S4632),
Hydroxyprogesterone caproate (S4674), Dichlorophen (S5724) all purchased from Selleck, Ulipristal acetate (MEC, HY-N0437) and Progesterone analogs all purchased from MCE. The antibodies used
in this study were obtained from the indicated source: rabbit anti-HA(CST, 3724; WB, 1:5,000), mouse anti-FLAG (Sigma, F3165; WB, 1:5,000), mouse anti-β-tubulin (ZENBIO, 200608; WB,
1:5,000), mouse anti-Myc (CST, 2276; WB, 1:5,000; IF, 1:500), goat anti-IL1β (R&D Systems, BAF401; WB, 1:2,000), rabbit anti-IL1β (Abcam, ab9722; WB, 1:5,000), rabbit anti-GSDMD (CST,
39754; WB: 1:2,000), rabbit anti-IL33 (Proteintech, 12372-1-AP; WB, 1:1,000), rabbit anti-V5 (CST, 13202; WB, 1:1,000; IF, 1:500), mouse anti-V5 (CST, 80076; WB, 1:5,000), rabbit anti-TMED10
(Proteintech, 15199-1-AP; WB, 1:3,000), rabbit anti-TMED2 (Proteintech, 11981-1-AP; WB, 1:3,000), mouse anti-GST (CST, 2624; WB, 1:5,000), rabbit anti-ERGIC53 (Sigma, E1031; WB, 1:2,000),
rabbit anti-GFP (CST, 2956; WB, 1:5,000), rabbit anti-Caspase3 (CST, 9662; WB: 1:2,000), rabbit anti-RPN1 (raised against C-terminal peptides corresponding to human protein residues 588–605
and mouse protein residues 576–605; from R. Schekman; WB, 1:5,000) and rabbit anti-E-SARS2 (raised against C-terminal peptides corresponding to the last 25 residues of E-SARS2 protein;
purified by ABclonal; WB, 1:1,000). Goat anti-rabbit IgG Alexa Fluor 568 (Invitrogen, A-11011), goat anti-mouse IgG Alexa Fluor 568 (Invitrogen, A-11004) and goat anti-mouse IgG Alexa Fluor
Plus 647 (Invitrogen, A32733) were used at a dilution of 1:500 for IF. MICE Mice were housed in ventilated cages kept at relatively stable temperature and humidity (20 °C-26 °C, 40%-70%) and
light regulated room (12 h light/12 h dark) in a SPF facility and received food and water ad libitum. C57BL/6 J mice were purchased from the Laboratory Animal Resources Center at Tsinghua
University. TMED10 fl/fl mice (C57BL/6) were created by GemPharmatech Co. Ltd, China. TMED10 inducible whole body knockout mice were generated via crossbreeding of TMED10 fl/fl with Cre-ERT
mice (from Dr. Xiaoyu Hu). 8-week-old male mice were intraperitoneally injected with tamoxifen (80 mg/kg) or corn oil for five continuous days. After the final tamoxifen injection, the mice
were fed normally for an additional week before being utilized in an AAV infection experiment. TRANSFECTION, LENTIVIRAL TRANSDUCTION AND SECRETION DETERMINATION Transfection of DNA
constructs into cells was performed using PEI (Polysciences, 23966) for HEK293T and X-tremeGENE HP (Roche, 6366244001) for U2OS according to the manufacturer’s protocols. Lentiviral
transduction was used to express TMED10, E, and mutants, GFP and GFP-ECT in THP-1, BEAS-2B or BMDMs. pLX304 plasmids containing the TMED10-V5 or FUGW plasmids containing Myc-E of indicated
coronavirus together with pMD2.G and psPAX2 were transfected into HEK293T cells to produce lentiviral particles for 72 h. The supernatant was collected to infect the indicated cells. For
secretion determination, cells were replaced with DMEM for 1 h or induced with 50 ng/ml LPS overnight in RPMI-1640 plus 10% FBS followed by 2 mM ATP treatment for 30 min in physiological
saline solution (147 mM NaCl, 10 mM HEPES pH 7.4, 13 mM glucose, 2 mM CaCl2, 1 mM MgCl2, 2 mM KCl). The medium was concentrated by an Amicon filter (Millipore, UFC5010) and cell lysate was
collected. Immunoblot was performed to determine the amounts of cargoes in the medium and cell. LDH assay (Thermo, 88953) was performed according to the manufacturer’s protocol.
IMMUNOFLUORESCENCE AND FLUORESCENCE COMPLEMENTATION For immunofluorescence of cultured cells, the cells were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature,
permeabilized with 0.1% TritonX-100 diluted in PBS at room temperature for 5 min, blocked with 10% FBS diluted with PBS for 1 h and primary antibody incubation for 1 h. After performing
multiple washes, the samples were incubated for 40 min at room temperature with the secondary antibodies74. For lung tissue immunofluorescence staining, samples were fixed in 4% PFA,
dehydrated in a 30% sucrose solution for 24 h, and embedded using the Tissue-Tek OCT compound. Frozen blocks were cut into 10-μm-thick sections. Fluorescence images were acquired using the
Olympus FV3000 confocal microscope. Quantification was performed using ImageJ. For fluorescence complementation, the cells expressing GFP(1-10)-TMED10-V5 and IL1β-FLAG-GFP11 were transfected
with E plasmids of indicated coronaviruses. The GFP signal in the cells was collected by CytoFlex LX (Beckman) and analyzed by CytExpert software17. CO-IMMUNOPRECIPITATION AND IN VITRO
PEPTIDE/GST PULL-DOWN ASSAY Co-immunoprecipitation assay was performed according methods reported previously75. The cells were lysed on ice for 30 min in IP buffer (50 mM Tris/HCl pH 7.4,
150 mM NaCl, 1 mM EDTA, 0.5% NP40, 10% glycerol) with protease inhibitor mixture, and the lysates were cleared by centrifugation. The resulting supernatants were incubated with indicated
agaroses and rotated at 4 °C for 3 h. Then the agaroses were washed five times with IP buffer followed by immunoblot. For peptide pull-down assay, synthetic peptides were conjugated to
agarose beads using the AminoLink Plus Coupling Resin (Thermo, 20501) according to the manufacturer’s protocol. 2 mg purified MBP-Myc-E protein of SARS2 was incubated with 20 μL
peptides-coupled beads in IP buffer and rotated at 4 °C for 3 h. Then the agarose was washed three times with IP buffer followed by immunoblot. For GST pull-down assay, the proteins were
purified and GST or GST-TMED10 was incubated with Glutathione agarose (GE, 17-0756-05) (which was blocked by 10% FBS) in IP buffer used for Co-IP, and rotated at 4 °C for 1 h. Then the beads
loaded with GST or GST-TMED10 were collected and incubated with MBP-Myc-E protein of SARS2 at 4 °C for 2 h. The beads were washed 3 times followed by immunoblot. CROSSLINK ASSAY DSS
crosslink assays were performed according to the instructions of the reagents. The cells were suspended in PBS with the 0.25 mM DSS at room temperature for 30 min. The reaction was quenched
with 20 mM Tris followed by sample preparation for immunoblot. PROTEIN PURIFICATION For protein purification, pGEX4T1-GST-TMED10, pGEX4T1-GST-Myc-E, pet28a-MBP-Myc-E, pet28a-mIL1β-flag and
pGEX4T1-GST-LgBit plasmids were transformed into E.coli Rosetta (DE3), cultured at 37 °C until OD600 reach 0.5-0.8, IPTG (100 μΜ) induced protein expression in 22 °C for 5 h. After
expression, the bacteria were collected and lysed with 0.5 mg/ml lysozyme (MP Biomedicine, 100831) in lysis buffer (2×PBS, 10 mM imidazole for His protein purification or 50 mM Tris/HCl pH
8.0, 5 mM EDTA,150 mM NaCl, 10% glycerol for GST protein purification) plus 0.3 mM DTT and PMSF(100 μΜ) on ice for 30 min. Added 0.5% TritonX-100 to lysate, sonicated and centrifuged at
20,000 × g for 40 min. Incubated supernatants with Glutathione Agarose or Ni-NTA Agarose (GE, 17-5318-02) and rotated at 4 °C for 2 h. Washed the agarose with wash buffer contain 0.1%
Tween20 and then wash buffer (2×PBS + 25 mM imidazole for His protein or PBS for GST protein). For purification of TMED10 and E, 0.5% Triton X-100 was included in all procedures. The
proteins were eluted by elution buffer (2×PBS, 250 mM imidazole for His proteins or 50 mM Tris, pH 8.0, 250 mM KCl, 25 mM glutathione for GST proteins) and concentrated by Amicon Ultra
Filters (Millipore, UFC9010). FPLC was performed for buffer exchange and increase of purity. The proteins were frozen by liquid nitrogen and stored in PBS (0.05% Triton X-100 for TMED10 and
E) at -80 °C. IN VITRO TRANSLOCATION ASSAY Total lipids extracted from HEK293T cells. Cell suspension and chloroform/methanol solution (methanol: chloroform = 1: 2) were mixed with a ratio
1: 4 by volume, vortexed for 30 s and shaken for 1 h at 180 rpm at 37 °C. After centrifugation at 2000 × _g_ for 10 min at room temperature, chloroform phase was collected and was evaporated
by a stream of nitrogen gas over the lipid solution and further dried in 37 °C incubator for 1 h. Dried lipid was suspended in HEPES-KAc buffer (20 mM HEPES, pH 7.2, 150 mM KCl). The
phosphatidylcholine (PC) content of lipid solution was measured (Phospholipids C, Wako) and used as a standard to normalize lipid concentration. The lipid was aliquoted and stored in -80 °C.
For reconstitution of proteoliposomes, total lipids were frozen and thawed 10 times in liquid nitrogen and 42 °C water bath. Add 0.05% TritonX-100 into lipid solution and rotated in 4 °C
for 30 min. TMED10 and E proteins were added into the lipid solution (10 μg GST-TMED10 protein, 10 μg Myc-E protein and 1.25 mg lipid in each tube) and incubated for another 1 h with
rotation. Each 400 μL solution was incubated with 6–8 mg Biobeads SM2 (Bio-Rad) equilibrated with the HEPES-KAc buffer at 4 °C. Beads were replaced each hour and repeated for 5 times (10 mg
beads in the third time and incubated overnight). After a 1500 × _g_ centrifugation to remove the Biobeads, the liposome solution was repeatedly frozen in liquid nitrogen and thawed in 42 °C
water bath for 5 times. In order to remove the free proteins, a membrane flotation procedure was performed. For each 300 μL solution, 300 μL 50% OptiPrep (diluted in HEPES-KAc buffer) was
added. The mixture was overlaid with 480 mL 20% OptiPrep and 90 μl HEPES-KAc buffer, centrifuged at 45,000 rpm (TLS55) for 2 h at 4 °C and the 150 μL top fraction (which contains the
proteoliposomes) was collected and diluted with 150 mM HEPES-KAc buffer. For the in vitro translocation, IL1β protein was added to proteoliposomes (150 μL reaction system contain 6 μg IL1β),
incubated for 1 h at 30 °C. After then, an equal volume of 50% OptiPrep (diluted in HEPES-KAc buffer) was added and mixed gently followed by overlaying with 240 μL 20% OptiPrep, 45 μL
HEPES-KAc buffer, and centrifuged at 45,000 rpm (TLS55) for 2 h at 4 °C. The proteoliposomes (90 μL fraction from the top) were aliquoted into 3 fractions. The first fraction was a control,
the second and third fractions were digested by protease K (15 μg/ml) without or with 0.5% Triton X-100 for 20 min on ice. The reactions were stopped by 1 mM PMSF and incubated for 10 min on
ice. Then SDS loading buffer was added and the samples were heated at 100 °C for 10 min followed by immunoblot analysis. CELL-FREE TRANSLOCATION ASSAY The cytosol of wild-type HEK293T cells
was prepared followed the research methods reported in the previous literature76. The cells were harvested and washed with PBS followed by passing through a 22 G needle in B88 lysis buffer
(20 mM HEPES-KOH, pH 7.2, 250 mM sorbitol, 150 mM potassium acetate, 5 mM magnesium acetate, protease and phosphotase inhibitors, 0.3 mM DTT). The cell homogenates were centrifuged at
100,000 × _g_ for 30 min, after which the supernatant fractions were collected and stored in -80 °C. For cytosol containing GFP or GFP-ECT, HEK293T cells were transfected with the indicated
plasmids. For membrane separation, the cells transfected with plasmids expressing TMED10 or different E proteins were harvested and lysed in HB1 lysis buffer (20 mM HEPES-KOH, pH 7.2, 400 mM
sucrose, 1 mM EDTA, protease and phosphotase inhibitors, 0.3 mM DTT) by using a 22 G needle. The lysate was centrifuged at 3000 × _g_ for 10 min and the supernatant was ultracentrifuged at
100,000 × _g_ for 30 min to collect the membrane pellet. The pellet was washed with B88 and resuspended in B88 lysis buffer containing the cytosol of wild-type HEK293T cells (3 mg/ml final
concentration). The phosphatidylcholine (PC) concentration was measured with a microplate spectrophotometer and adjusted to the equal concentration. For the cell-free mIL1β translocation,
recombinant proteins (120 μL reaction system contain 10 μg T7-mIL1β-flag protein), ATP regeneration system (40 mM creatine phosphate, 0.2 mg/ml creatine phosphokinase, and 1 mM ATP) and GTP
(0.15 mM) were added to the purified membrane solution containing 3 mg/ml cytosol, then incubated for 1.5 h in 30 °C. After then, 100 μL reaction system was combined with 200 μL 60% OptiPrep
to adjust the final concentration of OptiPrep to 40%, followed by overlaying with 600 μL 30% OptiPrep (diluted in B88 buffer), 100 μL B88 buffer, and centrifuged at 45,000 rpm (TLS55) for 2
h at 4 °C. The membrane fraction floating on the top (150 μL) was collected and aliquoted into three fractions. The first fraction was a control, the second and third fractions were
digested by protease K (20 μg/ml) without or with 1% Triton X-100 for 20 min on ice, with a total volume of 40 μL per reaction. The reactions were stopped by adding PMSF and incubated for 10
min on ice. Then SDS loading buffer was added and the samples were heated at 100 °C for 10 min followed by immunoblot analysis. AAV INFECTION AND LPS CHALLENGE The lung-tropic AAV serotype
6 vector expressing GFP, E-SARS2, E-229E and GFP-3×ECT under the control of a CMV promoter were generated by BraninVTA Co. Ltd, China. AAVs were delivered to lung using intratracheal
injection technique38. In brief, mice were anesthetized with avertin and a 22-gauge catheter placed into the trachea, then a total of 1 × 1011 vector genomes (vg) AAVs were administered.
Four weeks later, mice received an LPS challenge (15 mg/kg) and, 15 hours following the challenge, were euthanized to collect blood serum, lung, spleen, liver, and kidney for analysis using
ELISA, immunofluorescence, RT-qPCR and H&E staining. RNA EXTRACTION AND RT-QPCR Total RNA was extracted using Trizol (Beyotime, R0016). Reverse transcription to produce cDNA was carried
out by use a cDNA Reverse Transcription kit (Abclonal, RK20429), which contained 1 μg RNA in 20 μL volume. RT-qPCR was carried out by the 2×SYBR Green Master Mix (Abclonal, RK21203). Primers
sequences of IL6 and GAPDH are as follows: IL6-F: TGTATGAACAACGATGATGCACTT, IL6-R: ACTCTGGCTTTGTCTTTCTTGTTATCT; GAPDH-F: GTTCCTACCCCCAATGTGTCC, GAPDH-R: TAGCCCAAGATGCCCTTCAGT. H&E STAIN
AND INFLAMMATION EVALUATION Left lobe of mice lung were fixed in 4% paraformaldehyde, paraffin embedded, and sectioned (7 μm). Tissue sections were stained with hematoxylin and eosin. The
severity of lung inflammation was quantified by the percentage of inflammatory area: the area of the regions which immune cell filling to the lung alveolus and increased thickness dividing
total area of lung (excluding the air space)77. MHV INFECTION Mouse hepatitis virus A59 strain (MHV-A59) was propagated and tilter was determined according to the protocol78. In brief, the
virus was propagated in 17Cl-1 cells, subjected to three freeze-thaw cycles between -80 °C and room temperature, centrifuged at 5000 × _g_ for 20 min at 4 °C to remove cell debris, and the
supernatant was then concentrated using an Amicon filter. The tilter of the virus was determined by endpoint dilution assay78. In the cell infection assay, 293T-mCC1a cells were firstly
transfected with mIL1β plasmids and cultured for 24 h, then infected with MHV-A59 for 2 h in a serum-free medium, and subsequently cultured in a complete medium containing 10% FBS and 1%
Pan-Strep for 36 h. The culture medium was then switched to DMEM for 1 h before collecting the medium to assess mIL1β secretion as described above. To evaluate the impact of drugs, BMDMs
were infected with MHV-A59 (MOI 0.1), cultured for 36 h, treated with either DMSO or specific drugs for 4 h in serum-free medium, and the medium was subsequently collected to measure
endogenous mIL1β secretion. To examine the impact of ECT, BMDMs underwent lentiviral infection to express either GFP or GFP-ECT and were cultured for 48 h. These cells were then infected
with MHV-A59 (MOI 0.1), followed by assessment of endogenous IL1β secretion. For mice infection assay, 8–12 weeks old IFNAR-KO mice (from Dr. Fuping You) were intranasally inoculated with
MHV-A59 (2 × 104 PFU) using isoflurane as the anesthetic. The weight and health status of the mice were observed and recorded daily. Mice were euthanized at 4 dpi to collect serum and
different tissues for further testing. To assess drug efficacy, mice were first infected with MHV and then received intraperitoneal injections of DMSO, UPA (1 mg/kg), or Progesterone (1
mg/kg) for three consecutive days, with euthanasia occurring on the fourth day post-infection. Regarding AAV infection, mice underwent initial infection with either AAV-GFP or AAV-GFP-ECT,
followed by MHV infection four weeks later. COMPOUND SCREENING Rapid secretion analysis assay using complementary NanoLuc leuciferace was employed to screen inhibitors for E-SARS2-induced
mIL1β secretion53. 100 nL of compounds and DMSO or H2O controls were added to 96-well plate by Echo 650 Liquid Handler (Beckman). HEK293T cell expression mIL1β-HiBiT and Myc-E-SARS2 were
seeded to a 96-well plate, and cultured at 37 °C for 8 h. The medium was collected and each 30 μL medium incubated with Furimazine (10 nM final concentration) and 5 ng LgBit protein in a 60
μL reaction system at room temperature for 10 min. Luminescence was detected using EnSpire Multimode Plate Reader (PerkinElmer). Compounds with fluorescence intensity decreased by more than
0.75-fold relative to control in two independent experiments were selected as candidates. STATISTICS AND REPRODUCIBILITY Statistics analyzes were performed using GraphPad Prism 8.0.
Micrographs in Fig. 2a, b, and Supplementary Fig S4a are representatives of three independent experiments and were acquired randomly. All blot data are representative of at least three
independent experiments. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY All data
supporting the findings of this study are presented in the paper, Supplementary Information or Source Data file. All data and strains are available upon request. Source data are provided
with this paper. REFERENCES * Tay, M. Z., Poh, C. M., Rénia, L., MacAry, P. A. & Ng, L. F. The trinity of COVID-19: immunity, inflammation and intervention. _Nat. Rev. Immunol._ 20,
363–374 (2020). Article PubMed PubMed Central CAS Google Scholar * De Wit, E., Van Doremalen, N., Falzarano, D. & Munster, V. J. SARS and MERS: recent insights into emerging
coronaviruses. _Nat. Rev. Microbiol._ 14, 523–534 (2016). Article PubMed PubMed Central Google Scholar * Zhang, Y.-y, Li, B.-r & Ning, B.-t The comparative immunological
characteristics of SARS-CoV, MERS-CoV, and SARS-CoV-2 coronavirus infections. _Front. Immunol._ 11, 2033 (2020). Article PubMed PubMed Central CAS Google Scholar * Del Valle, D. M. et
al. An inflammatory cytokine signature predicts COVID-19 severity and survival. _Nat. Med._ 26, 1636–1643 (2020). Article PubMed PubMed Central Google Scholar * Xia, B. et al. SARS-CoV-2
envelope protein causes acute respiratory distress syndrome (ARDS)-like pathological damages and constitutes an antiviral target. _Cell Res._ 31, 847–860 (2021). Article PubMed PubMed
Central CAS Google Scholar * Yang, L. et al. COVID-19: immunopathogenesis and Immunotherapeutics. _Signal Transduct. Target. Ther._ 5, 128 (2020). Article PubMed PubMed Central CAS
Google Scholar * Hu, B., Huang, S. & Yin, L. The cytokine storm and COVID‐19. _J. Med. Virol._ 93, 250–256 (2021). Article PubMed CAS Google Scholar * Tan, M. et al.
Immunopathological characteristics of coronavirus disease 2019 cases in Guangzhou, China. _Immunology_ 160, 261–268 (2020). Article PubMed PubMed Central CAS Google Scholar * Qin, C. et
al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. _Clin. Infect. Dis._ 71, 762–768 (2020). Article PubMed CAS Google Scholar *
Mantovani, A., Dinarello, C. A., Molgora, M. & Garlanda, C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. _Immunity_ 50, 778-795,
https://doi.org/10.1016/j.immuni.2019.03.012 (2019). * Kaneko, N., Kurata, M., Yamamoto, T., Morikawa, S. & Masumoto, J. The role of interleukin-1 in general pathology. _Inflamm.
regeneration_ 39, 1–16 (2019). Article CAS Google Scholar * Griesenauer, B. & Paczesny, S. The ST2/IL-33 axis in immune cells during inflammatory diseases. _Front. Immunol._ 8, 475
(2017). Article PubMed PubMed Central Google Scholar * Markovic, S. S. et al. IL 33 correlates with COVID-19 severity, radiographic and clinical finding. _Front. Med._ 8, 749569 (2021).
Article Google Scholar * Conti, P. et al. Coronavirus-19 (SARS-CoV-2) induces acute severe lung inflammation via IL-1 causing cytokine storm in COVID-19: a promising inhibitory strategy.
_J. Biol. Regul. Homeost. Agents_ 34, 1971–1975 (2020). PubMed CAS Google Scholar * Makaremi, S. et al. The role of IL-1 family of cytokines and receptors in pathogenesis of COVID-19.
_Inflamm. Res._ 71, 923–947 (2022). Article PubMed PubMed Central CAS Google Scholar * Monteleone, M., Stow, J. L. & Schroder, K. Mechanisms of unconventional secretion of IL-1
family cytokines. _Cytokine_ 74, 213–218 (2015). Article PubMed CAS Google Scholar * Zhang, M. et al. A translocation pathway for vesicle-mediated unconventional protein secretion.
_Cell_ 181, 637–652.e615 (2020). Article PubMed CAS Google Scholar * Zhang, M. & Schekman, R. Unconventional secretion, unconventional solutions. _Science_ 340, 559–561 (2013).
Article ADS PubMed CAS Google Scholar * Evavold, C. L. et al. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. _Immunity_ 48, 35–44.e36
(2018). Article PubMed CAS Google Scholar * Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. _Nature_ 526, 666–671 (2015). Article ADS
PubMed CAS Google Scholar * Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. _Nature_ 526, 660–665 (2015). Article ADS PubMed CAS Google
Scholar * Chen, W. et al. Allergen protease-activated stress granule assembly and gasdermin D fragmentation control interleukin-33 secretion. _Nat. Immunol._ 23, 1021–1030 (2022). Article
PubMed PubMed Central CAS Google Scholar * Yang, L. et al. Intraepithelial mast cells drive gasdermin C-mediated type 2 immunity. _Immunity_ 57, 1056–1070.e1055 (2024). Article PubMed
CAS Google Scholar * Dupont, N. et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. _EMBO J._ 30, 4701–4711 (2011). Article PubMed PubMed Central
CAS Google Scholar * Martín-Sánchez, F. et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. _Cell Death Differ._ 23, 1219–1231 (2016). Article PubMed
PubMed Central Google Scholar * Monteleone, M. et al. Interleukin-1β maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and-independent secretion. _Cell
Rep._ 24, 1425–1433 (2018). Article PubMed CAS Google Scholar * Su, S. et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. _Trends Microbiol._ 24, 490–502
(2016). Article PubMed PubMed Central CAS Google Scholar * He, F., Deng, Y. & Li, W. Coronavirus disease 2019: What we know? _J. Med. Virol._ 92, 719–725 (2020). Article PubMed
PubMed Central CAS Google Scholar * Fung, T. S. & Liu, D. X. Human coronavirus: host-pathogen interaction. _Annu. Rev. Microbiol._ 73, 529–557 (2019). Article PubMed CAS Google
Scholar * Stertz, S. et al. The intracellular sites of early replication and budding of SARS-coronavirus. _Virology_ 361, 304–315 (2007). Article PubMed CAS Google Scholar * Schoeman,
D. & Fielding, B. C. Coronavirus envelope protein: current knowledge. _Virol. J._ 16, 1–22 (2019). Article CAS Google Scholar * Venkatagopalan, P., Daskalova, S. M., Lopez, L. A.,
Dolezal, K. A. & Hogue, B. G. Coronavirus envelope (E) protein remains at the site of assembly. _Virology_ 478, 75–85 (2015). Article PubMed CAS Google Scholar * Wang, Y. et al.
TMED10-mediated unconventional secretion of IL-33 regulates intestinal epithelium differentiation and homeostasis. _Cell Res._ 1-4 https://doi.org/10.1038/s41422-023-00891-3 (2024). * Zhang,
M., Kenny, S. J., Ge, L., Xu, K. & Schekman, R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. _elife_ 4, e11205 (2015). Article PubMed
PubMed Central Google Scholar * Vora, S. M., Lieberman, J. & Wu, H. Inflammasome activation at the crux of severe COVID-19. _Nat. Rev. Immunol._ 21, 694–703 (2021). Article PubMed
PubMed Central Google Scholar * Sun, X. et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. _Cell Death Differ._ 29, 1240–1254 (2022).
Article PubMed PubMed Central CAS Google Scholar * Ferreira, A. C. et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. _Cell death Discov._ 7, 43 (2021).
Article PubMed PubMed Central CAS Google Scholar * Santry, L. A. et al. AAV vector distribution in the mouse respiratory tract following four different methods of administration. _BMC
Biotechnol._ 17, 1–11 (2017). Article Google Scholar * Mandala, V. S. et al. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. _Nat.
Struct. Mol. Biol._ 27, 1202–1208 (2020). Article PubMed PubMed Central CAS Google Scholar * Surya, W., Li, Y., Verdià-Bàguena, C., Aguilella, V. M. & Torres, J. MERS coronavirus
envelope protein has a single transmembrane domain that forms pentameric ion channels. _Virus Res._ 201, 61–66 (2015). Article PubMed CAS Google Scholar * Verdiá-Báguena, C. et al.
Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. _Virology_ 432, 485–494 (2012). Article PubMed Google Scholar * Wang, Y. et al.
Impact of SARS-CoV-2 envelope protein mutations on the pathogenicity of Omicron XBB. _Cell Discov._ 9, 80 (2023). Article PubMed PubMed Central Google Scholar * Wang, W.-A.,
Carreras-Sureda, A. & Demaurex, N. SARS-CoV-2 infection alkalinizes the ERGIC and lysosomes through the viroporin activity of the viral envelope protein. _J. Cell Sci._ 136, jcs260685
(2023). Article PubMed PubMed Central CAS Google Scholar * Nieto-Torres, J. L. et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the
NLRP3 inflammasome. _Virology_ 485, 330–339 (2015). Article PubMed CAS Google Scholar * Yalcinkaya, M. et al. Modulation of the NLRP3 inflammasome by Sars-CoV-2 Envelope protein. _Sci.
Rep._ 11, 24432 (2021). Article ADS PubMed PubMed Central CAS Google Scholar * Nieto-Torres, J. L. et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel
activity promotes virus fitness and pathogenesis. _PLoS Pathog._ 10, e1004077 (2014). Article PubMed PubMed Central Google Scholar * Pervushin, K. et al. Structure and inhibition of the
SARS coronavirus envelope protein ion channel. _PLoS Pathog._ 5, e1000511 (2009). Article PubMed PubMed Central Google Scholar * Regla-Nava, J. A. et al. Severe acute respiratory
syndrome coronaviruses with mutations in the E protein are attenuated and promising vaccine candidates. _J. Virol._ 89, 3870–3887 (2015). Article PubMed PubMed Central CAS Google Scholar
* Hossain, A., Akter, S., Rashid, A. A., Khair, S. & Alam, A. R. U. Unique mutations in SARS-CoV-2 Omicron subvariants’ non-spike proteins: Potential impacts on viral pathogenesis and
host immune evasion. _Microb. pathogenesis_ 170, 105699 (2022). Article CAS Google Scholar * Shuai, H. et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron.
_Nature_ 603, 693–699 (2022). Article ADS PubMed CAS Google Scholar * Bálint, G., Vörös-Horváth, B. & Széchenyi, A. Omicron: increased transmissibility and decreased pathogenicity.
_Signal Transduct. Target. Ther._ 7, 151 (2022). Article PubMed PubMed Central Google Scholar * Nieto-Torres, J. L. et al. Subcellular location and topology of severe acute respiratory
syndrome coronavirus envelope protein. _Virology_ 415, 69–82 (2011). Article PubMed CAS Google Scholar * Schwinn, M. K. et al. CRISPR-mediated tagging of endogenous proteins with a
luminescent peptide. _ACS Chem. Biol._ 13, 467–474 (2018). Article PubMed CAS Google Scholar * Ghosh, S. et al. β-Coronaviruses use lysosomes for egress instead of the biosynthetic
secretory pathway. _Cell_ 183, 1520–1535.e1514 (2020). Article PubMed PubMed Central CAS Google Scholar * Guo, Q. et al. Induction of alarmin S100A8/A9 mediates activation of aberrant
neutrophils in the pathogenesis of COVID-19. _Cell host microbe_ 29, 222–235.e224 (2021). Article PubMed CAS Google Scholar * Sharma, L. et al. Distinct roles of type I and type III
interferons during a native murine β coronavirus lung infection. _J. Virol._ 96, e01241–01221 (2022). Article PubMed PubMed Central CAS Google Scholar * Yuan, L. et al. Female sex
hormone, progesterone, ameliorates the severity of SARS-CoV-2-caused pneumonia in the Syrian hamster model. _Signal Transduct. Target. Ther._ 7, 47 (2022). Article PubMed PubMed Central
CAS Google Scholar * Su, S. et al. Modulation of innate immune response to viruses including SARS-CoV-2 by progesterone. _Signal Transduct. Target. Ther._ 7, 137 (2022). Article PubMed
PubMed Central CAS Google Scholar * Zhou, S. et al. SARS-CoV-2 E protein: Pathogenesis and potential therapeutic development. _Biomed. Pharmacother._, 114242 (2023). * Schoeman, D. &
Fielding, B. C. Is there a link between the pathogenic human coronavirus envelope protein and immunopathology? A review of the literature. _Front. Microbiol._ 11, 2086 (2020). Article
PubMed PubMed Central Google Scholar * Zheng, M. et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. _Nat. Immunol._ 22, 829–838 (2021). Article PubMed
PubMed Central CAS Google Scholar * De Maio, F. et al. Improved binding of SARS-CoV-2 Envelope protein to tight junction-associated PALS1 could play a key role in COVID-19 pathogenesis.
_Microbes Infect._ 22, 592–597 (2020). Article PubMed PubMed Central Google Scholar * Chai, J. et al. Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction
protein PALS1. _Nat. Commun._ 12, 3433 (2021). Article ADS PubMed PubMed Central CAS Google Scholar * Lin, X. et al. Unconventional secretion of unglycosylated ORF8 is critical for
the cytokine storm during SARS-CoV-2 infection. _PLoS Pathog._ 19, e1011128 (2023). Article PubMed PubMed Central CAS Google Scholar * Matsuoka, K. et al. SARS-CoV-2 accessory protein
ORF8 is secreted extracellularly as a glycoprotein homodimer. _J. Biol. Chem._ 298, 101724 (2022). * Lin, X. et al. ORF8 contributes to cytokine storm during SARS-CoV-2 infection by
activating IL-17 pathway. _Iscience_ 24, 102293 (2021). * Miao, G. et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for
autolysosome formation. _Developmental cell_ 56, 427–442.e425 (2021). Article PubMed CAS Google Scholar * Ji, M. et al. VMP1 and TMEM41B are essential for DMV formation during
β-coronavirus infection. _J. Cell Biol._ 221, e202112081 (2022). Article PubMed PubMed Central Google Scholar * Trimarco, J. D. et al. TMEM41B is a host factor required for the
replication of diverse coronaviruses including SARS-CoV-2. _PLoS Pathog._ 17, e1009599 (2021). Article PubMed PubMed Central CAS Google Scholar * Schneider, W. M. et al. Genome-scale
identification of SARS-CoV-2 and pan-coronavirus host factor networks. _Cell_ 184, 120–132.e114 (2021). Article PubMed CAS Google Scholar * Mukherjee, S. & Pahan, K. Is COVID-19
gender-sensitive? _J. Neuroimmune Pharmacol._ 16, 38–47 (2021). Article PubMed PubMed Central Google Scholar * Zhang, J. et al. A systemic and molecular study of subcellular localization
of SARS-CoV-2 proteins. _Signal Transduct. Target. Ther._ 5, 269 (2020). Article ADS PubMed PubMed Central CAS Google Scholar * Hu, X. et al. Integrated regulation of Toll-like
receptor responses by Notch and interferon-γ pathways. _Immunity_ 29, 691–703 (2008). Article PubMed PubMed Central CAS Google Scholar * Li, S. et al. A new type of ERGIC–ERES membrane
contact mediated by TMED9 and SEC12 is required for autophagosome biogenesis. _Cell Res._ 32, 119–138 (2022). Article PubMed Google Scholar * Ma, X. et al. CCT2 is an aggrephagy receptor
for clearance of solid protein aggregates. _Cell_ 185, 1325–1345.e1322 (2022). Article PubMed CAS Google Scholar * Ge, L., Melville, D., Zhang, M. & Schekman, R. The ER–Golgi
intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. _elife_ 2, e00947 (2013). Article PubMed PubMed Central Google Scholar *
Lopera, D. et al. Structural and topographic dynamics of pulmonary histopathology and local cytokine profiles in Paracoccidioides brasiliensis conidia-infected mice. _PLoS Neglected Tropical
Dis._ 5, e1232 (2011). Article CAS Google Scholar * Leibowitz, J., Kaufman, G. & Liu, P. Coronaviruses: propagation, quantification, storage, and construction of recombinant mouse
hepatitis virus. _Curr. Protoc. Microbiol._ 21, 15E. 11.11–15E. 11.46 (2011). Article Google Scholar Download references ACKNOWLEDGEMENTS We thank Dr. Feng Shao (National Institute of
Biological Sciences, China) for GSDMD-KO mice, Dr. Fuping You (Peking University, China) for IFNAR-KO mice and MHV-A59. We thank Dr. Xiaoyu Hu (Tsinghua University, China) for the assistant
of experimental technique. We thank Dr. Zhaobing Gao and Dr. Bingqing Xia (Chinese Academy of Sciences, China) for suggestions on this project. We thank Dr. Ziyi Guo, Dr. Fengwu Chen, Qing
Guo, Xiangyue Mu for the help of mice experiment. This work is funded by National Natural Science Foundation of China (32370728), Vanke Special Fund for Public Health and Health Discipline
Development, Tsinghua University (2022Z82WKJ009), National Natural Science Foundation of China (92254302, 32225013, 32200553, 32130023), Tsinghua University Dushi Program, Tsinghua
University Initiative Scientific Research Program (No.20221080048, No.20231080030). AUTHOR INFORMATION Author notes * These authors contributed equally: Lei Liu, Lijingyao Zhang. AUTHORS AND
AFFILIATIONS * State Key Laboratory of Membrane Biology, Tsinghua University, Beijing, 100084, China Lei Liu, Lijingyao Zhang, Xinyan Hao, Yang Wang, Liang Ge & Min Zhang * School of
Pharmaceutical Sciences, Tsinghua University, Beijing, 100084, China Lei Liu, Xinyan Hao, Xiaochun Zhang, Boxue Tian & Min Zhang * Tsinghua University-Peking University Joint Center for
Life Sciences, School of Life Sciences, Tsinghua University, Beijing, 100084, China Lijingyao Zhang, Yang Wang & Liang Ge * Key Laboratory for Experimental Teratology of Ministry of
Education and Advanced Medical Research Institute, Meili Lake Translational Research Park, Cheeloo College of Medicine, Shandong University, Jinan, 250012, China Peihui Wang Authors * Lei
Liu View author publications You can also search for this author inPubMed Google Scholar * Lijingyao Zhang View author publications You can also search for this author inPubMed Google
Scholar * Xinyan Hao View author publications You can also search for this author inPubMed Google Scholar * Yang Wang View author publications You can also search for this author inPubMed
Google Scholar * Xiaochun Zhang View author publications You can also search for this author inPubMed Google Scholar * Liang Ge View author publications You can also search for this author
inPubMed Google Scholar * Peihui Wang View author publications You can also search for this author inPubMed Google Scholar * Boxue Tian View author publications You can also search for this
author inPubMed Google Scholar * Min Zhang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS M.Z. and L.G. conceived the project. L.L., L.Z.,
P.W., and M.Z. performed cell and biochemical experiments. L.L., L.Z., and Y.W. performed AAV infection experiments. L.L. and L.Z. performed drug screening experiments. X.Z and B.T
performed the bioinformatics analysis to search the analogs of progesterone. L.L., L.Z., and X.H. performed the MHV infection experiments. L.L., L.Z., and M.Z. analyzed the data. M.Z., L.G.,
L.L., and L.Z. wrote the manuscript. CORRESPONDING AUTHOR Correspondence to Min Zhang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER
REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION
PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed
under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material.
You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the
article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Liu, L., Zhang, L., Hao, X. _et al._ Coronavirus envelope protein activates
TMED10-mediated unconventional secretion of inflammatory factors. _Nat Commun_ 15, 8708 (2024). https://doi.org/10.1038/s41467-024-52818-0 Download citation * Received: 30 March 2024 *
Accepted: 20 September 2024 * Published: 08 October 2024 * DOI: https://doi.org/10.1038/s41467-024-52818-0 SHARE THIS ARTICLE Anyone you share the following link with will be able to read
this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Salman rushdie's 'feisty and defiant' humour is intact, says sonNew York: Salman Rushdie is still in a critical condition, but his usual feisty and defiant sense of humour remains inta...
Face-to-face with PHANGS | Nature AstronomyAccess through your institution Buy or subscribe The multiwavelength views present different, but complementary, impress...
Steven gerrard has doubts about duo ahead of rangers’ match against hibernianRangers duo Connor Goldson and Juninho Bacuna are both doubts with knocks for the top of the cinch Premiership table cla...
Poll shows print newspaper readership on the decline - aarp bulletinAn era in which Americans read a daily newspaper with their morning coffee, or on the subway on their way to work, is fa...
Family 'heartbroken' after 'harmless' man stabbed to death in 'race hate crime'The family of a "kind-hearted" and "harmless" man who died after being attacked in a 'race-hate...
Latests News
Coronavirus envelope protein activates tmed10-mediated unconventional secretion of inflammatory factorsABSTRACT The precise cellular mechanisms underlying heightened proinflammatory cytokine production during coronavirus in...
What is a Community Vision? - GOV.UK* Guidance WHAT IS A COMMUNITY VISION? Updated 27 January 2025 CONTENTS * The role of the GDF Community Partnership * Wh...
England age group assessment routes confirmed for 2023-24 | england hockeyThe ENGLAND AGE GROUP squads are the pinnacle of the England Hockey TALENT SYSTEM and assessment includes: * Players fro...
Boston city council district 8 race: sharon durkan and montez haywoodPolitics TWO CANDIDATES ARE RUNNING FOR DISTRICT 8: INCUMBENT SHARON DURKAN AND MONTEZ HAYWOOD. We surveyed both candida...
A clearer picture of microbial biogeographyThis month’s Under the Lens discusses recent advances in spatial metabolomics and light-sheet microscopy for imaging the...