Tapasin assembly surveillance by the rnf185/membralin ubiquitin ligase complex regulates mhc-i surface expression
Tapasin assembly surveillance by the rnf185/membralin ubiquitin ligase complex regulates mhc-i surface expression"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Immune surveillance by cytotoxic T cells eliminates tumor cells and cells infected by intracellular pathogens. This process relies on the presentation of antigenic peptides by Major
Histocompatibility Complex class I (MHC-I) at the cell surface. The loading of these peptides onto MHC-I depends on the peptide loading complex (PLC) at the endoplasmic reticulum (ER).
Here, we uncovered that MHC-I antigen presentation is regulated by ER-associated degradation (ERAD), a protein quality control process essential to clear misfolded and unassembled proteins.
An unbiased proteomics screen identified the PLC component Tapasin, essential for peptide loading onto MHC-I, as a substrate of the RNF185/Membralin ERAD complex. Loss of RNF185/Membralin
resulted in elevated Tapasin steady state levels and increased MHC-I at the surface of professional antigen presenting cells. We further show that RNF185/Membralin ERAD complex recognizes
unassembled Tapasin and limits its incorporation into PLC. These findings establish a novel mechanism controlling antigen presentation and suggest RNF185/Membralin as a potential therapeutic
target to modulate immune surveillance. SIMILAR CONTENT BEING VIEWED BY OTHERS STRUCTURE OF AN MHC I–TAPASIN–ERP57 EDITING COMPLEX DEFINES CHAPERONE PROMISCUITY Article Open access 14
September 2022 UBIQUITIN-LIKE PROTEIN 3 (UBL3) IS REQUIRED FOR MARCH UBIQUITINATION OF MAJOR HISTOCOMPATIBILITY COMPLEX CLASS II AND CD86 Article Open access 11 April 2022 VIRAL IMMUNE
EVASINS IMPACT ANTIGEN PRESENTATION BY ALLELE-SPECIFIC TRAPPING OF MHC I AT THE PEPTIDE-LOADING COMPLEX Article Open access 27 January 2022 INTRODUCTION The biogenesis of secretory and
membrane proteins in the endoplasmic reticulum (ER) is monitored by ER-associated degradation (ERAD)1,2. This conserved quality control system detects misfolded, unassembled, and
mislocalized proteins and promotes their transport back to the cytosol for degradation by the proteasome. ERAD also targets some folded proteins in a signal-dependent manner, such as
specific enzymes involved in sterol biosynthesis, thereby playing central roles in both protein and lipid homeostasis1,3. Mechanistically, ERAD is carried out by a variety of ubiquitin
ligase complexes integral to the ER membrane, each with specificity for different classes of substrates4. In all cases, subunits of ERAD complexes recognise substrates in the lumen or
membrane of the ER and facilitate their movement across the ER membrane toward the cytosol for ubiquitination. All ERAD complexes converge on the cytosolic p97 ATPase complex that pulls
ubiquitinated substrates from the membrane and hands them to the proteasome for degradation2,5. An ERAD complex consisting of the ubiquitin ligase RNF185, the multispanning membrane protein
Membralin (MBRL), and a member of the TMUB family—either TMUB1 or TMUB2—was recently identified6,7. We previously showed that this RNF185/MBRL complex promotes the degradation of a subset of
membrane proteins7. However, the complete set of RNF185/MBRL substrates remains unknown. Interestingly, genetic studies showed that MBRL ablation is perinatal lethal in mice8. While
indistinguishable from WT littermates at birth, MBRL KO mice display acute loss of motor neurons and hyperreactive astrocytes, resulting in death around day 5. Astrocyte-specific ablation of
MBRL resulted in a similar albeit milder phenotype, with the lethality occurring around day 259. Despite being ubiquitously expressed, the lethality of MBRL deletion was rescued by
expression of a neuronal MBRL transgene, indicating an essential role for this protein in the central nervous system, perhaps in astrocytes. How the severe mouse phenotype relates to MBRL
function in ERAD is unclear. Among the complexes assembled in the ER of vertebrate cells is the peptide loading complex (PLC). The PLC transports antigenic peptides generated in the cytosol
into the ER lumen and loads them onto major histocompatibility complex class I (MHC-I) molecules. Once loaded, MHC-I molecules traffic to the plasma membrane and display the peptides at the
cell surface for immune recognition by cytotoxic T-cells. These events are at the heart of adaptive immunity and are critical in the elimination of virally infected and cancerous cells10,11.
Import of peptides into the ER lumen depends on TAP1 and TAP2, ATP-binding cassette transporters embedded in the ER membrane12,13,14. Another component of the PLC is Tapasin (TPSN)15, which
recruits MHC-I to the PLC by binding to TAP1/2 via its transmembrane segment16,17,18,19, and to MHC-I via its large luminal domain20. A luminal loop of TPSN also performs an editing
function, ensuring that a high-affinity peptide binds the highly polymorphic groove on MHC-I21,22,23. TPSN has reduced affinity for peptide-loaded MHC-I molecules allowing them to dissociate
from the PLC and traffic to the cell surface21. The functions of TPSN, TAP1 and TAP2 at the PLC are assisted by the luminal chaperones ERp57 and Calreticulin24,25. While the assembly, MHC-I
loading, and editing at the PLC have been studied in detail, quality control processes regulating PLC function have not been identified. Here, using an unbiased quantitative proteomics
screen in mouse astrocytes, we identify the core PLC subunit TPSN as a substrate of the RNF185/MBRL complex and uncover the molecular basis for its ERAD recognition. We show that the
RNF185/MBRL complex ensures that TPSN functions exclusively in the peptide loading complex. Loss of this ERAD-mediated fail-safe mechanism in RNF185 or MBRL-deficient cells results in
increased MHC-I surface levels in professional antigen-presenting cells. Our findings highlight how the exquisite substrate specificity of ERAD is harnessed to ensure accurate antigen
presentation. Considering the importance of antigen presentation in diseases such as cancer, our results suggest that TPSN regulation by ERAD may be exploited therapeutically to modulate
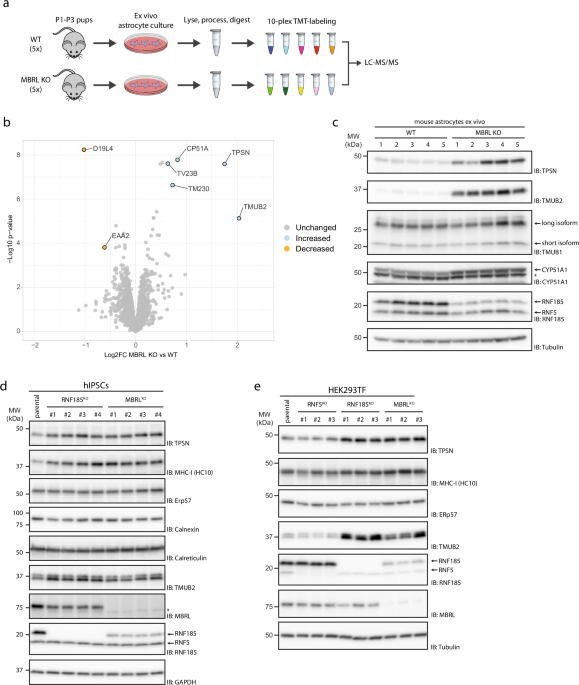
immune surveillance. RESULTS TAPASIN LEVELS ARE INCREASED IN RNF185/MBRL-DEFICIENT CELLS To gain insight into the physiological role of the RNF185/MBRL ERAD complex, we sought to identify
its endogenous substrates in mouse astrocytes, a cell type where MBRL function appeared to be critical8,9. We reasoned that endogenous substrates of this ERAD complex would be present at
higher steady-state levels in MBRL KO cells. Therefore, whole-cell quantitative proteomics was used to compare protein abundance in astrocytes derived from MBRL KO mice with their WT
littermates (Fig. 1a). CYP51A1 and TMUB2, two previously identified substrates of the RNF185/MBRL complex7, were among the most increased proteins in MBRL KO astrocytes confirming the
suitability of this approach for the identification of novel substrates (Fig. 1b, Source Data 2). Another highly enriched protein in MBRL KO astrocytes was Tapasin (TPSN), a single-spanning
ER membrane protein essential for loading antigenic peptides into MHC-I molecules26,27. Western blotting analysis confirmed these results (Fig. 1c). Consistent with the experiments in mouse
astrocytes, deletion of MBRL in human induced pluripotent stem cells (iPSCs) (Fig. 1d), HEK293 (Fig. 1e), THP-1 (Supplementary Fig. 1a), and U2OS (Supplementary Fig. 1b) cells also resulted
in higher TPSN steady-state levels. Importantly, the deletion of the MBRL-binding partner RNF185 in these cells resulted in a similar increase in TPSN steady-state levels (Fig. 1d, e).
Moreover, the effect was specific as the levels of other ER proteins, including the ER chaperone ERp57, which is also a TPSN-binding partner and critical for MHC-I presentation28, were
unchanged (Fig. 1d, e, Supplementary Fig. 1a, b). Deletion of the ubiquitin ligase RNF5, more than 70% identical to RNF18529, but unable to assemble with MBRL7, did not affect TPSN
steady-state levels in HEK293, THP-1, and U2OS cells. Therefore, ablation of the RNF185/MBRL ERAD complex results in a specific increase of TPSN steady-state levels in various cell types.
TAPASIN IS A SUBSTRATE OF THE RNF185/MBRL ERAD COMPLEX The observations described above suggested that TPSN is a substrate of the RNF185/MBRL ERAD complex. To investigate this possibility
further, we generated a TPSN transgene fused to sfGFP and HA-tags (TPSN-GFP-HA) expressed from a tetracycline responsive promotor. TPSN KO cells display a severe defect in MHC-I cell surface
expression15,30. Expression of TPSN-GFP-HA in TPSN KO HEK293 cells restored MHC-I surface levels indicating that the fusion protein is functional (Fig. 2a). In agreement with these results,
TPSN-GFP-HA assembled with its partners at the peptide loading complex as detected by immunoprecipitation followed by mass spectrometry (Fig. 2b, Source Data 3). Interestingly, TPSN also
showed robust association with many ERAD factors, including MBRL. The association of TPSN with the RNF185/MBRL ERAD complex was independently confirmed by western blotting (Fig. 2c).
Together these data showed that transgenic TPSN behaves like the endogenous protein and is a suitable tool to investigate TPSN regulation. The steady-state levels of TPSN-GFP-HA were
increased upon acute inhibition of the p97 ATPase or the proteasome, indicating that TPSN-GFP-HA was being degraded by ERAD (Supplementary Fig. 2a, b). Deletion of RNF185 or MBRL also
resulted in higher steady-state levels of TPSN-GFP-HA, while deletion of the ERAD ligases RNF5 or HRD1 had little or no effect (Fig. 3a and Supplementary Fig. 2c). Importantly, re-expression
of WT RNF185 or MBRL in their respective knockout lines restored normal steady-state levels of TPSN-GFP-HA. In contrast, re-expression of catalytically inactive RNF185 failed to rescue TPSN
levels, indicating that the ubiquitin ligase activity is essential in controlling TPSN levels (Supplementary Fig. 2d, e). Depletion of TMUB1/2 (Supplementary Fig. 2f–h), part of the
RNF185/MBRL complex, and UBE2K, the conjugating enzyme working with this complex, also resulted in increased steady-state levels of TPSN-GFP-HA (Supplementary Fig. 2i). To analyse TPSN
turnover we performed chase experiments upon translation shut-off with cycloheximide. These experiments confirmed that, in parental cells, TPSN-GFP-HA was a short-lived protein with a
half-life of approximately 4 h (Fig. 3b). Importantly, degradation of TPSN-GFP-HA was blocked in RNF185 and MBRL KO cells while ablation of RNF5 did not affect TPSN turnover (Fig. 3b).
Degradation of ERAD substrates requires their prior ubiquitination1,4. We tested whether RNF185/MBRL stimulated TPSN degradation by promoting its ubiquitination. Consistent with its short
half-life, TPSN ubiquitin-conjugates were readily detected in parental cells. (Fig. 3c). In contrast, RNF185 or MBRL KO cells showed much lower levels of ubiquitinated TPSN, even if their
overall levels were higher in the KO cells. This effect was specific because cells deficient in the ERAD ubiquitin ligases RNF5 or HRD1 displayed levels of ubiquitinated TPSN comparable to
controls (Fig. 3c). There are four lysine residues within the short cytosolic tail of TPSN. Lysines are the most common acceptor sites for ubiquitination during protein degradation. We
tested the importance of these lysine residues in TPSN degradation by mutating them to alanine in the TPSN 4K-to-A tail. This mutant showed increased steady-state levels compared to WT TPSN
and was largely resistant to an acute ERAD block upon p97 inhibition (Fig. 3d). Moreover, the TPSN 4K-to-A tail mutant was only poorly ubiquitinated (Fig. 3e), while its association with the
RNF185/MBRL complex was unaffected (Supplementary Fig. 2j). Therefore, lysine residues in TPSN cytosolic tail are important for its ubiquitination but not for its binding to the RNF185/MBRL
ERAD complex. Altogether, these data indicate that TPSN steady-state levels are regulated by RNF185/MBRL-mediated ERAD. We previously observed that among the RNF185/MBRL complex components,
MBRL appeared to be the one that co-precipitated most efficiently with CYP51A1TM, another substrate of this ERAD complex7. This suggested that MBRL could play a role in selecting substrates
for degradation. To test this possibility further, we asked whether MBRL could interact with TPSN independently of the other subunits of the RNF185/MBRL complex. Indeed, we observed that
endogenous MBRL could precipitate TPSN in cells lacking RNF185 and TMUB1/2 (Fig. 3f). This association was specific as only background levels of TPSN were associated with the ERAD ligase
HRD1. Therefore, MBRL binds TPSN independently of other components of the complex and likely plays an important role in TPSN recognition. Previous studies showed that the poxvirus Molluscum
contagiosum downregulates MHC-I surface expression of the host cells by promoting the rapid degradation of TPSN by an ERAD-like process31. This process requires the virally encoded protein
MC80. We then tested whether MC80 was promoting TPSN degradation by hijacking the RNF185/MBRL ERAD complex. In agreement with previous studies31, we observed that expression of MC80 resulted
in lower TPSN steady-state levels that could be rescued upon inhibition of p97 or the proteasome (Supplementary Fig. 3a). However, deletion of RNF185 or MBRL did not interfere with
MC80-induced degradation of TPSN (Supplementary Fig. 3b, c). Consistently, the ubiquitin-conjugating enzyme UBE2K, which assists RNF185/MBRL-dependent ubiquitination7 was also dispensable
for MC80-induced degradation of TPSN. In contrast, depletion of ubiquitin-conjugating UBE2G2 (Supplementary Fig. 3d) or the cognate ERAD ligases HRD1 and GP78 (Supplementary Fig. 3e)
resulted in inhibition of TPSN degradation induced by MC80. Altogether, these data indicate that normally, TPSN degradation depends on the RNF185/MBRL complex, while in cells expressing the
Molluscum contagiosum protein MC80, TPSN is degraded by distinct ERAD complexes. CHARGED RESIDUE IN THE MEMBRANE DOMAIN DETERMINES TAPASIN ASSEMBLY AND DEGRADATION Next, we investigated the
conditions leading to TPSN degradation by the RNF185/MBRL complex. Our prior analysis did not identify any other components of the PLC as interactors of the RNF185/MBRL complex7. Therefore,
we raised the possibility that RNF185/MBRL was interacting with and promoting the degradation of TPSN that was not assembled with its PLC partners. To test this possibility, we analysed TPSN
levels in cells lacking TAP1, a TPSN partner at the PLC that transports antigenic peptides into the ER lumen for loading into the MHC-I15. Consistent with earlier observations32,
steady-state levels of TPSN were reduced in TAP1-deficient cells. Strikingly, deletion of RNF185 or MBRL in TAP1 KO cells restored normal TPSN levels suggesting that unassembled TPSN was
indeed a substrate of the RNF185/MBRL complex (Fig. 4a and Supplementary Fig. 4a). The effect was specific as simultaneous deletion of TAP1 and the ubiquitin ligase RNF5 had little or no
effect on TSPN levels. One interpretation of these results is that TPSN assembly with PLC components and its degradation by the RNF185/MBRL complex are competing events. Consistent with this
idea, we observed that overexpression of TAP1 resulted in increased TPSN steady-state levels (Fig. 4b) and stability (Fig. 4c). Interestingly, the turnover of overexpressed TAP1 occurs
independently of MBRL (Fig. 4c). Collectively, these data indicate that RNF185/MBRL promotes the degradation of unassembled TPSN and that once assembled with other PLC components, TPSN is no
more degraded by ERAD. Next, we investigated how unassembled TPSN was recognized by the RNF185/MBRL complex. TPSN assembles with TAP1 and TAP2 via its transmembrane segment16,17,18,19.
Specifically, conserved positively charged lysine and negatively charged aspartate residues in TPSN and TAP1/2, respectively, form an intramembrane salt bridge important for the assembly of
these proteins in the PLC16 (Fig. 4d). We then asked whether these assembly determinants of TPSN were also important for its degradation by ERAD. To directly test this possibility, we
generated soluble TPSN (sTPSN), a TPSN mutant lacking the C-terminal portion including the transmembrane segment27, and TPSN K428A, with a single point mutation in the conserved
intramembrane lysine residue at position 428. These proteins were expressed at high levels, and importantly, their steady-state levels were insensitive to p97 inhibition or deletion of the
RNF185/MBRL complex, indicating that these TPSN mutants were not degraded by ERAD (Supplementary Fig. 4b, c). Indeed, cycloheximide chase experiments confirmed that in contrast to wt TPSN,
TPSN K428A is a stable protein (Fig. 4e). Moreover, TPSN K428A failed to interact with the RNF185/MBRL complex consistent with it not being an ERAD substrate (Fig. 4f). Thus, the conserved
intramembrane lysine at position 428 functions both as critical determinant of TPSN assembly into the PLC and as a degradation signal for the RNF185/MBRL ERAD complex. The dual role of the
lysine 428 in assembly and degradation ensures that TPSN is functional only within the PLC, with molecules failing to assemble being quickly degraded in an RNF185/MBRL-dependent manner.
Seminal work on the assembly of the T-cell receptor (TCR) revealed the importance of intramembrane-charged residues in membrane protein quality control33. Specifically, the degradation of
unassembled TCR alpha chain (TCR-α) was shown to depend on the presence of two charged residues (lysine and arginine) within its transmembrane segment34,35. Given the parallels with our
findings with TPSN, we asked whether RNF185/MBRL also played a role in the recognition of the charges within TCR-α that led to its ERAD. To this end, TCR-α transmembrane segment (TCR-α TM)
was expressed as a fusion to the interleukin-2 receptor a chain (Tac antigen) as before (Supplementary Fig. 4d)33 and its steady-state levels were assessed in various ERAD mutants
(Supplementary Fig. 4e). Interestingly, steady-state levels of TCR-α TM were unaffected by RNF185/MBRL or RNF5 mutations but increased upon depletion of the ubiquitin ligase HRD1. These data
suggest that while K428 is essential for TPSN recognition, RNF185/MBRL does not have a general role in the recognition of intramembrane charges during quality control. We also analysed
CYP26A1 TM, a model ERAD substrate identified by our laboratory and that consists of the transmembrane segment of CYP26A1 (Sergejevs et al., in revision). Despite lacking intramembrane
charges (Supplementary Fig. 4d), CYP26A1 TM degradation requires RNF185/MBRL complex and is unaffected by depletion of RNF5 or HRD1 (Supplementary Fig. 4e). Therefore, intramembrane-charged
residues may be essential for ERAD of some substrates, but they are not sufficient to determine specificity for an individual ERAD complex. Moreover, these data highlight the complexity of
substrate recognition during membrane protein quality control. INCREASED MHC-I SURFACE EXPRESSION UPON LOSS OF RNF185/MBRL COMPLEX The vital role of TPSN in MHC-I antigen presentation
prompted us to test if its regulation by the RNF185/MBRL complex impacted the cell surface levels of MHC-I. To this end, we focused on macrophages, which are professional antigen-presenting
cells, derived from human iPSCs. Parental, RNF185 and MBRL KO iPSC lines presented in Fig. 1d were differentiated into macrophages using a well-established protocol36. As observed in other
cell types, loss of RNF185 or MBRL resulted in higher TPSN steady-state levels (Fig. 5a). Intriguingly, the increase in TPSN levels appeared more prominent in macrophages than in the
corresponding iPSCs from where they were derived (compare Figs. 1d and 5a). The mutant macrophages also showed increased total levels of MHC-I as well as the TPSN partners at the PLC, TAP1
and TAP2. The increase was more pronounced for TAP1 than TAP2, in agreement with earlier observations showing the interdependence of the steady-state levels of TPSN, TAP1 and TAP218,32. In
contrast, the levels of several ER chaperones, including the TPSN-binding partner ERp57, were indistinguishable between parental and RNF185 or MBRL KO macrophages indicating that the effects
observed were specific (Fig. 5a). Upregulation of components of the antigen presentation pathway such as MHC-I and the PLC normally occurs upon activation of JAK-STAT signalling, either
upon infection or stimulation of cells with the interferon gamma (IFNγ) cytokine37. Nevertheless, the increased levels of TPSN in untreated RNF185 or MBRL KO macrophages was not a
consequence of constitutive activation of JAK-STAT signalling as monitored by the phosphorylation status of STAT1 tyrosine at position 701 (pSTAT1; Fig. 5a). Moreover, stimulation of the
cells with IFNγ resulted in upregulation of PLC components and MHC-I as well as STAT1 phosphorylation, indicating that JAK-STAT signalling is functional in RNF185 or MBRL KO macrophages.
Importantly, RNF185 or MBRL KO macrophages showed higher steady-state levels of TPSN in relation to controls, even after IFNγ stimulation. Finally, we used a conformational sensitive
antibody that detects peptide-loaded MHC-I molecules to assess the cell surface levels of MHC-I. Both under resting and IFN-stimulated conditions, loss of RNF185 or MBRL KO macrophages
showed elevated surface levels of peptide-MHC-I complexes (Fig. 5b). The effects were specific for MHC-I, as the levels of cell surface proteins CD14 and CD45 were comparable between the
RNF185 or MBRL KOs and control cells. Thus, the RNF185/MBRL ERAD complex regulates MHC-I surface expression. DISCUSSION The PLC is critical for the loading of antigenic peptides onto MHC-I
molecules and adaptive immunity. While the process of peptide loading onto MHC-I by the PLC has been extensively studied, quality control mechanisms acting directly on PLC components are
unknown. Here we identified an ERAD-based surveillance mechanism that regulates the levels of the core PLC component TPSN and that is important for MHC-I surface expression. We showed that
TPSN molecules that are not assembled with their PLC partners become substrates of the RNF185/MBRL ERAD complex. This process depends on the binding of RNF185/MBRL to the transmembrane
segment of unassembled TPSN. Importantly, this binding involves an evolutionarily conserved lysine residue (K428) within the TPSN transmembrane domain that is also essential for its assembly
in the PLC. In this case, K428 forms an intramembrane salt bridge with conserved, negatively charged aspartate residues of its partners TAP1 or TAP2 (Fig. 6). Thus, both assembly and
degradation of TPSN depend on K428. This mechanism is reminiscent of the one described for the assembly and quality control of the T-cell receptor (TCR)33,34,35,38. In this case, charged
residues within the transmembrane segment of the TCR-α chain important for assembly with the CD3-δ subunit are also critical to trigger degradation of unassembled TCR-α33,34. While
intramembrane-charged residues are common assembly determinants for a variety of protein complexes, how they are recognized by quality control factors is less clear. For example, RNF185/MBRL
recognizes the intramembrane positively charged residue in TPSN, but not the ones in TCR-α. This highlights the exquisite specificity of the various ERAD complexes and suggests that
additional determinants are involved in conferring their substrate specificity. The higher TPSN levels observed upon loss of RNF185/MBRL manifested in increased surface expression of MHC-I
in iPSC-derived macrophages. Concomitant with the rise in TPSN levels we observed an increase in the levels of TAP1 and TAP2, consistent with earlier observations27. The overall increase in
these core PLC components may enhance peptide loading onto MHC-I, resulting in higher levels of peptide-MHC-I complexes at the cell surface as observed in human iPSC-derived macrophages
deficient for RNF185/MBRL. Besides recruiting MHC-I to the PLC, TPSN favours the loading of high-affinity peptides onto MHC-I through a process known as “peptide editing”39,40. The increase
in TPSN levels likely reshapes the repertoire of antigenic peptides by favouring the loading of MHC-I with high-affinity peptides, which are more stable at the cell surface41,42. Increased
MHC-I surface expression in RNF185/MBRL-deficient macrophages may also result from reduced turnover. These two possibilities are not mutually exclusive and future studies should clarify
their individual contribution to the higher MHC-I surface expression observed in RNF185 and MBRL KO macrophages. Both the levels and the repertoire of antigenic peptides presented by MHC-I
are critical in eliciting cytotoxic T-cell responses43,44,45,46. Therefore, future studies should also assess if the changes in antigen presentation upon loss of RNF185/MBRL result in
abnormal recognition by cytotoxic T cells and are eventually exploited therapeutically to modulate immune responses. Earlier genetic studies revealed that an astrocyte-specific MBRL KO mouse
developed a neuroinflammatory-like phenotype that resulted in motor neuron death9. In vitro, studies attributed this phenotype to the accumulation of extracellular glutamate through
reducing the glutamate transporter EAAT2. While our proteomics analysis confirmed the reduction in EAAT2 levels, we wonder whether the dysregulated antigen presentation due to increased TPSN
levels also contributes to the MBRL KO mouse phenotype. An increase in MHC-I presentation has been linked to neuroinflammation in different settings47,48. Therefore, future studies should
test this possibility directly. METHODS Ethical approval for the mouse experiment was provided by the ethical committee at the Sanford Burnham Prebys Medical Discovery Institute. CELL LINES
U2OS and THP-1 cells were obtained from the ECACC. The Lenti-X 293T cell line for the production of lentivirus was obtained from TakaraBio. Flp-In T-REx HEK293 cells were obtained from
Invitrogen (Thermo Fischer Scientific). Flp-In T-REx HEK293 lines were established either as a clonal line or a polyclonal population according to the manufacturer’s guidelines. Cells were
grown at 37 °C 5% CO2 in DMEM medium (Merck Life Science UK Limited #D6429) supplemented with L-Glutamine (2 mM; Gibco #25030024), Penicillin-Streptomycin (10 Units/mL; Gibco #15140122) and
10% FCS (Merck Life Science UK Limited #F9665). THP-1 cells were grown in RPMI medium (Merck Life Science UK Limited #R8758) supplemented with l-Glutamine, Penicillin-Streptomycin, and 10%
FCS. PLASMIDS The pcDNA5-FRT-TO and pOG44 plasmids were obtained from Invitrogen. cDNAs or sgRNAs, respectively, for protein overexpression and gene deletions, were cloned in a dual promoter
lentiviral vector, as described previously49. The lentiviral TAP1 expression plasmid was kindly gifted by Emmanuel Wiertz. LENTIVIRUS PRODUCTION For gene transductions using lentiviruses,
the virus was produced using Lenti-X 293T cells in 24-well plates using TransIT LT1 (Mirus Bio LLC #MIR 2305) and second-generation packaging vectors pMD2.G and psPAX2 according to standard
lentiviral production protocols. GENERATION OF CRISPR/CAS9-MEDIATED KNOCKOUT CELLS For CRISPR/Cas9-mediated knockouts, cell lines were transfected using Mirus LT-1 according to the
manufacturer’s protocol. On the next day, cells were selected using Puromycin (2 μg/mL; Gibco #A1113803). After 48 h of selection, the selection medium was replaced with complete medium. To
generate KO clones, cells were single-cell sorted using a BD FACSAria3 or AriaFusion. The knockout status of the clones was confirmed via flow cytometry and immunoblotting. To generate
knockouts in THP-1 cells, cells were transduced in the presence of polybrene (Santa Cruz Biotechnology #28728-55-4) with lentivirus carrying CRISPR/Cas9. Two days after transduction, cells
were put on 2 μg/mL Puromycin selection for 7 days. SgRNA sequences used are listed in Supplementary Table 1. MICE All the mice were maintained in group housing on a 12-h light-dark cycle at
21 +/− 1 degree Celsius with ~50% humidity. Animals were given ad libitum access to food and water. The membralinfl/fl line was generated by inserting loxP sites in the introns preceding
exon 2 and following exon 4 by Dr. Dongxian Zhang at Sanford Burnham Prebys Medical Discovery Institute (SBP) (The Jackson Laboratory #016574). Membralin homozygous KO animals were generated
by crossing membralinfl/fl animals with Tg (_ACTB-Cre_) (The Jackson Laboratory #003376) to achieve germ-line deletion of membralin exons 2 to 4. Membralin heterozygous KO animals were
crossed to maintain the colony and generate homozygous KO animals (mem-KO). The sex of the animals was not selected. PRIMARY ASTROCYTE CULTURE Primary cortical astrocytes were prepared from
P1–P3 pups. Cortical tissue was treated with papain (80 U/mL; Worthington, cat# LS003126) for 20 min at 37 °C in an incubator and centrifuged for 5 min at 300 × _g_. Tissue pellets were
resuspended in DMEM/F12 media supplemented with 10% Fetal Bovine Serum (FBS) (Nacalai USA, cat# 174012-500ML-BULK) and 1% penicillin/streptomycin (Thermo-Fisher Scientific, cat# 15140122)
and mechanically dissociated using 1000 μL tips. Cells were seeded and cultured on 10 cm tissue culture-treated plates pre-coated with Matrigel (Corning, cat# 356237) for 21 days. The medium
was changed every other day. Prior to harvesting the astrocyte protein samples, microglia were removed by shaking the plates at 100 rpm overnight in the incubator. The astrocytes were
dissociated by 2.5% Trypsin (Thermo-Fisher Scientific, cat# 15090046) and washed in PBS for 5 times before being stored in −80 °C freezer. PROTEOME SAMPLE PREPARATION FROM EX VIVO MOUSE
ASTROCYTE CULTURE Mouse astrocytes were lysed in 5% SDS 50mM TEAB lysis buffer (pH 7.55). A total of 50 µg protein was reduced by incubation with 5 mM tris(2-carboxyethyl)phosphine (TCEP)
for 15 min at 37 °C, and subsequently alkylated with 20 mM iodoacetamide for 30 min at room temperature in the dark. Protein digestion was performed using the suspension trapping (S-Trap™)
sample preparation method using the manufacturer’s guidelines (ProtiFi™, Huntington NY). Briefly, 2.5 µL of 12% phosphoric acid was added to each sample, followed by the addition of 165 µL
S-Trap binding buffer (90% methanol in 100 mM TEAB, pH 7.1). This was added to the S-Trap Micro spin column. The samples were centrifuged at 4000 × _g_ for 2 min until all the solution
passed through the filter. Each S-Trap Mini-spin column was washed with 150 µL S-trap binding buffer by centrifugation at 4000 × _g_ for 1 min. This process was repeated for a total of four
washes. 25 µL of 50 mM TEAB, pH 8.0, containing trypsin (1:10 ratio of trypsin to protein) was added to each sample, followed by proteolytic digestion for 3 h at 47 °C using a thermomixer
(Eppendorf) without shaking. Peptides were eluted with 50 mM TEAB pH 8.0 and centrifugation at 4000 × _g_ for 2 min. Elution steps were repeated using 0.2% formic acid and 0.2% formic acid
in 50% acetonitrile, respectively. The three eluates from each sample were combined and dried using a speed-vac before storage at −80 °C. TMT-10 PLEX LABELLING Each 15 µg protein digest was
resuspended in 25 µL 100 mM HEPES, pH 8.5. TMT-10 plex labelling (TMT lot number: UG287488) was carried out as per the manufacturer’s instructions. Samples were assigned to a TMT tag and 10
µL of the corresponding TMT tag was added per sample. Samples were incubated for 1 hr at room temperature. An aliquot corresponding to 0.5 µg was taken from each sample and pooled together
for ratio and labelling efficiency checks, prior to making the full pooled sample. The test pool was quenched with 0.69 µL of 5% hydroxylamine, incubated for 15 min at room temperature, and
dried using a speed-vac. The dried samples were cleaned up using C18 spin column as per the manufacturer’s guidelines (Thermo Scientific), and subsequently dried using a speed-vac. Peptides
were dissolved in 2% acetonitrile with 0.1% TFA, and the pooled sample was analysed for labelling efficiency and ratio check. For the ratio check, each sample (corresponding to a single TMT
channel) was normalised to the median of all samples within its pool. Each sample was quenched with 4 µL 5% hydroxylamine and incubated for 15 min, and subsequently, samples were pooled
together based on the scaling factors, which were calculated using the test pool. Samples were dried using a speed vac, cleaned using PierceTM Peptide Desalting Spin columns as per the
manufacturer’s guidelines (Thermo Scientific), and dried down again using a speed vac prior to offline high-performance liquid chromatography (HPLC) fractionation. OFFLINE HPLC FRACTIONATION
Peptides were resuspended in 55 µL ammonium formate, pH 8.0. Peptides were fractionated on a Basic Reverse Phase column (Gemini C18, 3 μm particle size, 110A pore, 3 mm internal diameter,
250 mm length, Phenomenex #00G-4439-Y0) on a Dionex Ultimate 3000 off-line LC system. All solvents used were HPLC grade (Rathburn). Peptides (40 μL) were loaded on the column for 1 min at
250 μL/min using 99% Buffer A (20 mM ammonium formate, pH = 8) and eluted for 40 min on a linear gradient from 1 to 90 % Buffer B (100% ACN). Peptide elution was monitored by UV detection at
214 nm. Fractions were collected every 60 s from 2 min to 38 min for a total of 36 fractions. Fractions were pooled using non-consecutive concatenation to obtain 18 pooled fractions (e.g.
pooled fraction 1: fraction 1 + 19). Each fraction was acidified to a final concentration of 1% TFA and dried using a speed vac. MASS SPECTROMETRY FOR TMT-10 PLEX SAMPLES Peptides were
dissolved in 2% acetonitrile with 0.1% TFA, and each sample was independently analysed on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo-Fisher Scientific), connected to a
UltiMate 3000 RSLCnano System (Thermo-Fisher Scientific). Peptides (~2 μg per fraction) were injected on an Acclaim PepMap 100 C18 LC trap column (100 μm ID × 20 mm, 3 μm, 100 Å) followed by
separation on an EASY-Spray nanoLC C18 column (75 μm ID × 50 cm, 2 μm, 100 Å) at a flow rate of 250 nl min−1. Solvent A was water containing 0.1% formic acid, and solvent B was 80%
acetonitrile containing 0.1% formic acid. The gradient used for the analysis of proteome samples was as follows: solvent B was maintained at 3% for 5 min, followed by an increase from 3 to
35% B in 120 min, 35–90% B in 0.5 min, maintained at 90% B for 4 min, followed by a decrease to 3% in 0.5 min and equilibration at 3% for 20 min. Mass spectrometric identification and
quantification were performed on an Orbitrap Fusion Tribrid mass spectrometer (Thermo-Fisher Scientific) operated in data-dependent, positive-ion mode. Full scan spectra were acquired in a
range from 375 m/z to 1500 m/z, at a resolution of 120,000, with a standard automated gain control (AGC) (Tune 3.3) and a maximum injection time of 50 ms. Precursor ions were isolated with a
quadrupole mass filter width of 0.7 m/z and CID fragmentation was performed in one-step collision energy of 35% and 0.25 activation Q. Detection of MS/MS fragments was acquired in the
linear ion trap in a rapid mode using a Top 3s method, with a standard AGC target and a maximum injection time of 50 ms. The dynamic exclusion of previously acquired precursor was enabled
for 60 s with a tolerance of +/-10 ppm. Quantitative analysis of TMT-tagged peptides was performed using FTMS3 acquisition in the Orbitrap mass analyser operated at 60,000 resolution, with a
standard AGC target and maximum injection time of 105 ms. HCD fragmentation on MS/MS fragments was performed in a one-step collision energy of 65% to ensure maximal TMT reporter ion yield
and synchronous-precursor-selection (SPS) was enabled to include 10 MS/MS fragment ions in the FTMS3 scan. All spectra were analysed using MaxQuant 1.6.10.43 and searched against SwissProt
_mus musculus_ fasta files (containing 25,350 database entries with isoforms, downloaded on 2021/03/10). Peak list generation was performed within MaxQuant and searches were performed using
default parameters and the built-in Andromeda search engine. Reporter ion MS3 was used for quantification and the additional parameter of quantitation labels with 10 plex TMT on N‐terminal
or lysine was included. The enzyme specificity was set to consider fully tryptic peptides, and two missed cleavages were allowed. Oxidation of methionine and N-terminal acetylation were
allowed as variable modifications. Carbamidomethylation of cysteine was allowed as a fixed modification. A protein and peptide false discovery rate (FDR) of less than 1% was employed in
MaxQuant. Reporter ion intensities were used for data analysis. Briefly, the data was filtered to remove proteins that matched to a contaminant or a reverse database, which were only
identified by site, which were not quantified in every sample, or which contained less than 2 unique peptides. Reporter ion intensity values were log2 transformed. Moderated _t_-tests, with
patients accounted for in the linear model, were performed using Limma, where proteins with _p_-value < 0.05 were considered as statistically significant. All analysis was performed using
R 3.6.2. CO-IMMUNOPRECIPITATION Cells were lysed in 1% DMNG (Anatrace #NG322) lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl) containing a complete EDTA-free protease inhibitor cocktail
(Roche #5056489001). Lysates were rotated for 60 min at 4 °C. Cell debris and nuclei were pelleted at 13,000 × _g_ for 20 min at 4 °C. Post-nuclear supernatants were incubated for 2 h with
anti-HA magnetic beads (Pierce, Thermo-Fisher Scientific). After four washes in 0.1% DMNG washing buffer, proteins were eluted in 1x sample buffer for 30 min at 37 °C. The eluate was
transferred to a new tube and subsequently reduced using DTT (Merck Life Science UK Limited #D9779). Immunoblotting was performed as described below. PROTEOME SAMPLE PREPARATION FOR TAPASIN
INTERACTORS Immunoprecipitated samples on beads were resuspended in 25 µL 1x SDS sample buffer (5% SDS, 50 mM TAEB, pH 7.55). Disulfide bonds were reduced using 20 mM TCEP for 15 min at 47
°C. Following this, cooled samples were alkylated using 20 mM CAA in the dark for 15 min, and a 10% volume of 12% phosphoric acid was added to acidify the samples. S-trap binding buffer (90%
methanol in 100 mM TAEB, pH 7.5) was added to acidified, denatured samples to a final volume of 190 µL and the resulting solution was loaded onto S-Trap micro spin columns (Protify), with a
maximum of 150 µL of sample per load. Loaded spin columns were centrifuged at 4000 × _g_ for 1 min, and this step was repeated until the entire sample was loaded onto a spin column. S-Trap
columns were washed 5x with S-trap binding buffer (90% methanol in 100 mM TAEB, pH 7.5), and the columns were moved onto 2 mL low protein binding Eppendorf tubes. To each S-trap column, 25
µL of digestion solution (50 mM TAEB, pH 8.0), containing 2 µg of Trypsin/Lys-C mix (Promega V5071) was added and loosely capped columns were incubated for 3 h at 47 °C on a ThermoMixer.
Peptides were eluted with 30 µL of 50 mM TAEB, followed by 30 µL of 0.2% formic acid and 40 µL of 50% acetonitrile in 0.2% formic acid. Peptides were dried for 4 h at 37 °C in a vacuum
centrifuge, and samples were stored at −80 °C until further analysis. MASS SPECTROMETRY FOR TAPASIN INTERACTORS ANALYSIS Peptides were dissolved in 2% acetonitrile containing 0.1%
trifluoroacetic acid, and each sample was independently analysed on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo-Fisher Scientific), connected to an UltiMate 3000 RSLCnano
System (Thermo-Fisher Scientific). Peptides (1 µg) were injected on a PepMap 100 C18 LC trap column (300 μm ID × 5 mm, 5 μm, 100 Å) followed by separation on an EASY-Spray nanoLC C18 column
(75 μm ID × 50 cm, 2 μm, 100 Å) at a flow rate of 250 nl min−1. Solvent A was water containing 0.1% formic acid, and solvent B was 80% acetonitrile containing 0.1% formic acid. The gradient
used for analysis of proteome samples was as follows: solvent B was maintained at 2% for 5 min, followed by an increase from 2 to 35% B in 120 min, 35–90% B in 0.5 min, maintained at 90% B
for 4 min, followed by a decrease to 3% in 0.5 min and equilibration at 2% for 10 min. The Orbitrap Fusion Lumos was operated in positive-ion data-dependent mode. The precursor ion scan
(full scan) was performed in the Orbitrap in the range of 400–1600 m/z with a resolution of 120,000 at 200 m/z, an automatic gain control (AGC) target of 4 × 105, and an ion injection time
of 50 ms. MS/MS spectra were acquired in the linear ion trap (IT) using Rapid scan mode after high-energy collisional dissociation (HCD) fragmentation. An HCD collision energy of 30% was
used, the AGC target was set to 1 × 104, and dynamic injection time mode was allowed. The number of MS/MS events between full scans was determined on-the-fly to maintain a 3 s fixed duty
cycle. Dynamic exclusion of ions within a ± 10 ppm m/z window was implemented using a 35 s exclusion duration. An electrospray voltage of 2.0 kV and capillary temperature of 275 °C, with
no sheath and auxiliary gas flow, was used. Data was analysed in Perseus v1.6.14.0. For pairwise comparison of two conditions (control vs Tapasin), two-sided _t_-test was performed (s0 =
0.1, FDR = 0.05, number of randomisations = 250). Data was visualised using scatter plot. SUBSTRATE UBIQUITINATION EXPERIMENTS Cells at around 80–90% confluency in a 6-well were lysed in
RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% Sodium Deoxycholate, 0.1% SDS) containing NEM (20 mM; Merck Life Science UK Limited #E3876) and cOmplete protease
inhibitor cocktail (Roche #5056489001). Lysates were rotated for 60 min at 4 °C. Cell debris and nuclei were pelleted at 13,000 × _g_ for 20 min at 4 °C. Post-nuclear supernatants were
incubated for 2 h with anti-HA magnetic beads (Thermo-Fisher Scientific #88837). After four washes in RIPA buffer, proteins were eluted in the sample buffer. The eluate was transferred to a
new tube and subsequently reduced using DTT. Immunoblotting was performed as described below. TRANSLATION SHUT-OFF EXPERIMENTS Cells were seeded in wells pre-coated with poly-L-lysine (Merck
Life Science UK Limited #P8920). The next day, cells were incubated with cycloheximide (100 mg/mL; Merck Life Science UK Limited #C7698) for the indicated time points, after which cells
were directly lysed in 1x sample buffer containing Benzonase (Merck Life Science UK Limited #E1014), cOmplete protease inhibitor cocktail (Roche #5056489001), and DTT. Lysates were incubated
for 30 min at 37 °C, after which proteins were separated by SDS–PAGE. Immunoblotting was performed as described below. IMMUNOBLOTTING Samples were incubated at 37 °C for 15 min, separated
by SDS–PAGE (Bio-Rad), and proteins were transferred to PVDF membranes (Bio-Rad). Membranes were probed with antibodies against indicated proteins. All antibodies used are listed in
Supplementary Table 1. Reactive bands were detected by ECL (Western Lightning ECL Pro, Perkin Elmer #NEL121001EA), and visualized using an Amersham Imager 600 (GE Healthcare Life Sciences).
Data quantification was performed using Image Studio software (Li-Cor), and graphs were plotted in GraphPad Prism v9.5. Representative images of three independent experiments are shown. FLOW
CYTOMETRY For surface staining of MHC-I, cells were detached using EDTA, resuspended in FACS buffer (2% FBS, 2 mM EDTA in PBS), and washed once. Cells were incubated with W6/32-APC (1:50;
Biolegend #311410) for 1 h at 4 °C. Next, cells were washed twice in FACS buffer and directly analysed using a BD LSRFortessa X-20 flow cytometer. For fluorescence measurement of GFP, cells
were trypsinized, resuspended in FACS buffer, and directly analysed on the BD X-20. FACS data was analysed using FlowJo v10. IPSC CULTURE Human iPSC line SFC840-03-0350
(https://ebisc.org/STBCi026-A) and KO clones were cultured in OXE8 medium as described previously36. Briefly, the iPSCs were cultured on hESC-qualified Geltrex (ThermoFisherScientific, Cat#
A1413302) coated plates and passaged using 0.5 mM EDTA in PBS. SNP-QCed frozen stocks were used, and the passage number was kept to within 3 passages from the QCed stock. 10 μM Rho kinase
inhibitor (ROCKi) Y-27632 (Abcam, Cat# ab120129) was added to the culture medium during the first 24 h after thaw. Cells were incubated at 37 °C, 5% CO2. GENERATION OF IPSC KNOCKOUTS iPSCs
were treated with ROCKi for 1 h prior to nucleofection. iPSCs were harvested using EDTA, pelleted, and resuspended in Buffer R (Invitrogen) at a concentration of 2.2E7 cells/mL. Cells were
mixed with Alt-R™ AsCas12a (Cpf1) Ultra (IDT) crRNA complexes and nucleofection enhancer (IDT). Nucleofection was carried out using a Neon MPK5000 electroporator (Invitrogen) with the
following parameters: ‘HiTrans’ 1400v, 20ms width, 1 pulse. Cells were then transferred to prewarmed ROCKi-containing OXE8 medium and allowed to recover. To grow iPSC KO clones, mitotically
inactivated CF1 MEFs (Millipore #PMEF-CFL) were seeded in MEF medium (Advanced DMEM (Gibco #12491), 10% FCS, GlutaMAX (Gibco #35050), 50 μM 2-Mercaptoethanol (Gibco #31350)) on 0.1% gelatin
(Sigma G1393) coated plates. The next day, a limiting dilution of iPSC lines were seeded on top of the MEFs in hES medium (KnockOut DMEM (Gibco #10829), 20% KnockOut Serum replacement (Gibco
#10828), Non-Essential Amino Acids (Gibco #11140), GlutaMAX (Gibco), 50 μM 2 Mercaptoethanol (Gibco), Pen/Strep (Gibco #15140-122), 5 ng/mL bFGF (R&D #234-FSE/CF)) supplemented with
ROCKi. 50% of hES medium was changed daily until colonies were ready for manual picking. KO clones were selected by Western blotting (Fig. 1d). Additionally, SNP-karyotyping was performed on
genomic DNA to verify genomic integrity, using Illumina array Infinium GSA-24v3-0. DIFFERENTIATION OF IPSCS INTO MACROPHAGES iPSCs were differentiated into macrophages using a protocol
described previously36, and described here in brief. To form embryoid bodies (EBs), 4 million iPSCs were seeded into an AggreWell 800 plate (STEMCELL Technologies, no. 34815) in EB medium
(containing BMP4, VEGF and SCF) and incubated for 4 days at 37 °C, 5% CO2, with daily feeding of 75% medium change with EB medium. After 4 days, EBs were lifted from the plate and passed
over a 40 μm cell strainer (Corning) to remove dead cells, before washing into a tissue culture plate with differentiation medium (containing IL-3 and M-CSF). EBs were used to set up two
“factories” in two T175 flasks with 20 mL differentiation medium. Factories were incubated at 37 °C, 5% CO2 and fresh 10 mL differentiation medium added weekly until macrophage precursors
(PreMac) started to be produced. PreMac cells (emerging into the supernatant) were collected weekly and a minimum of equal volumes of differentiation media to the volume removed were
replaced in the factory. PreMac were passed through a 40 μm cell strainer to obtain a single-cell suspension and plated in appropriately sized tissue culture plates for further culturing in
OXM macrophage medium (containing M-SCF) for a further 7 days, with a 50% medium change on day 4. FLOW CYTOMETRY OF IPSC-DERIVED MACROPHAGES Cells were washed with PBS and lifted by
incubation with Accutase (Gibco, #A11105-01). Then cells were stained in FACS buffer (PBS supplemented with 1% FBS, 10 μg/mL human-IgG (Sigma, no. I8640-100MG), and 0.01% sodium azide) and
compared with isotype controls with the same fluorophores from the same company. Antibodies used included CD45-FITC (Immunotools, Cat# 21270453), CD14-FITC (Immunotools, Cat# 21270143),
isotype-FITC (Immunotools, Cat# 21335013), W6/32-APC (Biolegend, Cat# 311400) and isotype-APC (Biolegend, Cat# 400220). Fluorescence was measured using the BD LSRFortessa X-20 (BD
Biosciences) and analysed using FlowJo version 10. STATISTICS AND REPRODUCIBILITY Western blot data was quantified using Image Studio software (Li-Cor) and graphs were plotted using Prism
(GraphPad). Representative images of at least three independent experiments are shown. Error bars represent the standard deviation, the measure of centre represents the mean. REPORTING
SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The raw proteomics dataset generated during
this study is available at PRIDE as PXD048728. The western blotting data generated during this study are available at Mendeley Data (https://doi.org/10.17632/vfd47jgr8t). Source data are
provided in this paper. MATERIALS AVAILABILITY All unique reagents generated in this study are available from the lead contact without restriction. Further information and requests for
reagents may be directed to and will be fulfilled by the lead contact, Pedro Carvalho ([email protected]). REFERENCES * Christianson, J. C., Jarosch, E. & Sommer, T.
Mechanisms of substrate processing during ER-associated protein degradation. _Nat. Rev. Mol. Cell Biol._ 24, 777–796 (2023). Article CAS PubMed Google Scholar * Christianson, J. C. &
Carvalho, P. Order through destruction: how ER-associated protein degradation contributes to organelle homeostasis. _EMBO J._ 41, e109845 (2022). Article CAS PubMed PubMed Central
Google Scholar * van den Boomen, D. J. H., Volkmar, N. & Lehner, P. J. Ubiquitin-mediated regulation of sterol homeostasis. _Curr. Opin. Cell Biol._ 65, 103–111 (2020). Article PubMed
Google Scholar * Krshnan, L., van de Weijer, M. L. & Carvalho, P. Endoplasmic reticulum-associated protein degradation. _Cold Spring Harb. Perspect. Biol._ 14, a041247 (2022). Article
CAS PubMed Google Scholar * Wu, X. & Rapoport, T. A. Mechanistic insights into ER-associated protein degradation. _Curr. Opin. Cell Biol._ 53, 22–28 (2018). Article CAS PubMed
PubMed Central Google Scholar * Fenech, E. J. et al. Interaction mapping of endoplasmic reticulum ubiquitin ligases identifies modulators of innate immune signalling. _eLife_ 9, e57306
(2020). Article CAS PubMed PubMed Central Google Scholar * van de Weijer, M. L. et al. Quality control of ER membrane proteins by the rnf185/membralin ubiquitin ligase complex. _Mol.
Cell_ 79, 768–781.e7 (2020). Article PubMed PubMed Central Google Scholar * Yang, B. et al. The critical role of membralin in postnatal motor neuron survival and disease. _eLife_ 4,
e06500 (2015). Article PubMed PubMed Central Google Scholar * Jiang, L.-L. et al. Membralin deficiency dysregulates astrocytic glutamate homeostasis leading to ALS-like impairment. _J.
Clin. Invest._ 129, 3103–3120 (2019). Article PubMed PubMed Central Google Scholar * Blum, J. S., Wearsch, P. A. & Cresswell, P. Pathways of antigen processing. _Annu. Rev. Immunol._
31, 443–473 (2013). Article CAS PubMed PubMed Central Google Scholar * Trowitzsch, S. & Tampé, R. Multifunctional chaperone and quality control complexes in adaptive immunity.
_Annu. Rev. Biophys._ 49, 135–161 (2020). Article CAS PubMed Google Scholar * Androlewicz, M. J., Anderson, K. S. & Cresswell, P. Evidence that transporters associated with antigen
processing translocate a major histocompatibility complex class I-binding peptide into the endoplasmic reticulum in an ATP-dependent manner. _Proc. Natl Acad. Sci. USA_ 90, 9130–9134 (1993).
Article ADS CAS PubMed PubMed Central Google Scholar * Shepherd, J. C. et al. TAP1-dependent peptide translocation in vitro is ATP dependent and peptide selective. _Cell_ 74, 577–584
(1993). Article CAS PubMed Google Scholar * Neefjes, J. J., Momburg, F. & Hämmerling, G. J. Selective and ATP-dependent translocation of peptides by the MHC-encoded transporter.
_Science_ 261, 769–771 (1993). Article ADS CAS PubMed Google Scholar * Ortmann, B. et al. A critical role for tapasin in the assembly and function of multimeric MHC class I-TAP
complexes. _Science_ 277, 1306–1309 (1997). Article CAS PubMed Google Scholar * Blees, A. et al. Assembly of the MHC I peptide-loading complex determined by a conserved ionic
lock-switch. _Sci. Rep._ 5, 17341 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Petersen, J. L. et al. A charged amino acid residue in the transmembrane/cytoplasmic
region of tapasin influences MHC class I assembly and maturation. _J. Immunol._ 174, 962–969 (2005). Article CAS PubMed Google Scholar * Raghuraman, G., Lapinski, P. E. & Raghavan,
M. Tapasin interacts with the membrane-spanning domains of both TAP subunits and enhances the structural stability of TAP1 x TAP2 Complexes. _J. Biol. Chem._ 277, 41786–41794 (2002). Article
CAS PubMed Google Scholar * Leonhardt, R. M., Abrahimi, P., Mitchell, S. M. & Cresswell, P. Three tapasin docking sites in TAP cooperate to facilitate transporter stabilization and
heterodimerization. _J. Immunol._ 192, 2480–2494 (2014). Article CAS PubMed Google Scholar * Bangia, N., Lehner, P. J., Hughes, E. A., Surman, M. & Cresswell, P. The N-terminal
region of tapasin is required to stabilize the MHC class I loading complex. _Eur. J. Immunol._ 29, 1858–1870 (1999). Article CAS PubMed Google Scholar * Jiang, J. et al. Structural
mechanism of tapasin-mediated MHC-I peptide loading in antigen presentation. _Nat. Commun._ 13, 5470 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Müller, I. K. et al.
Structure of an MHC I-tapasin-ERp57 editing complex defines chaperone promiscuity. _Nat. Commun._ 13, 5383 (2022). Article ADS PubMed PubMed Central Google Scholar * Hafstrand, I. et
al. Successive crystal structure snapshots suggest the basis for MHC class I peptide loading and editing by tapasin. _Proc. Natl Acad. Sci. USA_ 116, 5055–5060 (2019). Article ADS CAS
PubMed PubMed Central Google Scholar * Peaper, D. R., Wearsch, P. A. & Cresswell, P. Tapasin and ERp57 form a stable disulfide-linked dimer within the MHC class I peptide-loading
complex. _EMBO J._ 24, 3613–3623 (2005). Article CAS PubMed PubMed Central Google Scholar * Sadasivan, B., Lehner, P. J., Ortmann, B., Spies, T. & Cresswell, P. Roles for
calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. _Immunity_ 5, 103–114 (1996). Article CAS PubMed Google Scholar * Wearsch, P. A.
& Cresswell, P. Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. _Nat. Immunol._ 8, 873–881 (2007).
Article CAS PubMed Google Scholar * Lehner, P. J., Surman, M. J. & Cresswell, P. Soluble tapasin restores MHC class I expression and function in the tapasin-negative cell line .220.
_Immunity_ 8, 221–231 (1998). Article CAS PubMed Google Scholar * Dong, G., Wearsch, P. A., Peaper, D. R., Cresswell, P. & Reinisch, K. M. Insights into MHC class I peptide loading
from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. _Immunity_ 30, 21–32 (2009). Article PubMed PubMed Central Google Scholar * El Khouri, E., Le Pavec, G.,
Toledano, M. B. & Delaunay-Moisan, A. RNF185 is a novel E3 ligase of endoplasmic reticulum-associated degradation (ERAD) that targets cystic fibrosis transmembrane conductance regulator
(CFTR). _J. Biol. Chem._ 288, 31177–31191 (2013). Article CAS PubMed PubMed Central Google Scholar * Garbi, N. et al. Impaired immune responses and altered peptide repertoire in
tapasin-deficient mice. _Nat. Immunol._ 1, 234–238 (2000). Article CAS PubMed Google Scholar * Harvey, I. B., Wang, X. & Fremont, D. H. Molluscum contagiosum virus MC80 sabotages
MHC-I antigen presentation by targeting tapasin for ER-associated degradation. _PLoS Pathog._ 15, e1007711 (2019). Article CAS PubMed PubMed Central Google Scholar * de Waard, A. A. et
al. PAKC: a novel panel of HLA class I antigen presentation machinery knockout cells from the same genetic origin. _Eur. J. Immunol._ 51, 734–737 (2021). Article PubMed PubMed Central
Google Scholar * Manolios, N., Bonifacino, J. S. & Klausner, R. D. Transmembrane helical interactions and the assembly of the T cell receptor complex. _Science_ 249, 274–277 (1990).
Article ADS CAS PubMed Google Scholar * Bonifacino, J. S., Cosson, P. & Klausner, R. D. Colocalized transmembrane determinants for ER degradation and subunit assembly explain the
intracellular fate of TCR chains. _Cell_ 63, 503–513 (1990). Article CAS PubMed Google Scholar * Bonifacino, J. S., Cosson, P., Shah, N. & Klausner, R. D. Role of potentially charged
transmembrane residues in targeting proteins for retention and degradation within the endoplasmic reticulum. _EMBO J._ 10, 2783–2793 (1991). Article CAS PubMed PubMed Central Google
Scholar * Vaughan-Jackson, A. et al. Differentiation of human induced pluripotent stem cells to authentic macrophages using a defined, serum-free, open-source medium. _Stem Cell Rep._ 16,
3093 (2021). Article CAS Google Scholar * Zhou, F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. _Int. Rev. Immunol._ 28, 239–260
(2009). Article CAS PubMed Google Scholar * Bonifacino, J. S., Suzuki, C. K., Lippincott-Schwartz, J., Weissman, A. M. & Klausner, R. D. Pre-Golgi degradation of newly synthesized
T-cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. _J. Cell Biol._ 109, 73–83 (1989). Article CAS PubMed Google Scholar * Thomas, C. & Tampé, R.
MHC I assembly and peptide editing—chaperones, clients, and molecular plasticity in immunity. _Curr. Opin. Immunol._ 70, 48–56 (2021). Article CAS PubMed Google Scholar * van Hateren, A.
& Elliott, T. The role of MHC I protein dynamics in tapasin and TAPBPR-assisted immunopeptidome editing. _Curr. Opin. Immunol._ 70, 138–143 (2021). Article PubMed Google Scholar *
Thirdborough, S. M. et al. Tapasin shapes immunodominance hierarchies according to the kinetic stability of peptide-MHC class I complexes. _Eur. J. Immunol._ 38, 364–369 (2008). Article CAS
PubMed Google Scholar * Hein, Z. et al. Peptide-independent stabilization of MHC class I molecules breaches cellular quality control. _J. Cell Sci._ 127, 2885–2897 (2014). CAS PubMed
Google Scholar * van der Burg, S. H., Visseren, M. J., Brandt, R. M., Kast, W. M. & Melief, C. J. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC-peptide
complex stability. _J. Immunol._ 156, 3308–3314 (1996). Article PubMed Google Scholar * Dalchau, N. et al. A peptide filtering relation quantifies MHC class I peptide optimization. _PLoS
Comput. Biol._ 7, e1002144 (2011). Article CAS PubMed PubMed Central Google Scholar * Assarsson, E. et al. A quantitative analysis of the variables affecting the repertoire of T cell
specificities recognized after vaccinia virus infection. _J. Immunol._ 178, 7890–7901 (2007). Article CAS PubMed Google Scholar * Kaseke, C. et al. HLA class-I-peptide stability mediates
CD8+ T cell immunodominance hierarchies and facilitates HLA-associated immune control of HIV. _Cell Rep._ 36, 109378 (2021). Article CAS PubMed PubMed Central Google Scholar * Cebrián,
C. et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. _Nat. Commun._ 5, 3633 (2014). Article ADS PubMed Google Scholar * Grigg, J. B.
et al. Antigen-presenting innate lymphoid cells orchestrate neuroinflammation. _Nature_ 600, 707–712 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * van de Weijer, M.
L. et al. A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. _Nat. Commun._ 5, 3832 (2014). Article ADS PubMed Google Scholar
* Fernandes, H. J. R. et al. ER stress and autophagic perturbations lead to elevated extracellular α-snuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. _Stem Cell Rep._ 6,
342–356 (2016). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank P. Cresswell and L. Boyle for antibodies to TPSN and TAPBPR1, respectively, E. Wiertz for the
lentiviral TAP1 expression plasmid, T. Rapoport and T. Williams for reading the manuscript, and O. Dushek, T. Elliot and P. Klenerman for discussions. P.C. was supported by an investigator
award from the Wellcome Trust (223153/Z/21/Z). P.C. and M.L.v.d.W. were supported by a Research Grant from the BBSRC (BB/W001519/1). L.J. was supported by K99 AG066960 from the National
Institute of Aging. S.A.C. was supported by the James Martin 21st Century Research Foundation. This research was co-funded by grant awards to M.T. (Wellcome Trust Multi-User Equipment grant
(212947/Z/18/Z) and Investigator Award (215542/Z/19/Z)). R.J.K. and T.Y.H. received support from R01 AG062190 (NIA, NIH). AUTHOR INFORMATION Author notes * LuLin Jiang Present address: Altos
Labs-Bay Institute of Science, Redwood City, CA, USA * Maria Emilia Dueñas Present address: Telethon Kids Institute, Perth, Nedlands, WA, 6009, Australia * Tiaan Heunis Present address:
Immunocore Ltd, 92 Park Drive, Abingdon, OX14 4RY, UK * These authors contributed equally: Krishna Samanta, Nikita Sergejevs. AUTHORS AND AFFILIATIONS * Sir William Dunn School of Pathology,
University of Oxford, South Parks Road, Oxford, OX1 3RE, UK Michael L. van de Weijer, Krishna Samanta, Nikita Sergejevs, Tiaan Heunis, Sumana Sanyal, Sally A. Cowley & Pedro Carvalho *
Degenerative Diseases Program, Genetics, and Aging Research Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA, 92037, USA LuLin Jiang, Timothy Y. Huang & Randal J.
Kaufman * Biosciences Institute, Newcastle University, Framlington Place, Newcastle upon Tyne, NE2 4HH, UK Maria Emilia Dueñas & Matthias Trost * James and Lillian Martin Centre for
Stem Cell Research, Sir William Dunn School of Pathology, University of Oxford, Oxford, UK Sally A. Cowley Authors * Michael L. van de Weijer View author publications You can also search for
this author inPubMed Google Scholar * Krishna Samanta View author publications You can also search for this author inPubMed Google Scholar * Nikita Sergejevs View author publications You
can also search for this author inPubMed Google Scholar * LuLin Jiang View author publications You can also search for this author inPubMed Google Scholar * Maria Emilia Dueñas View author
publications You can also search for this author inPubMed Google Scholar * Tiaan Heunis View author publications You can also search for this author inPubMed Google Scholar * Timothy Y.
Huang View author publications You can also search for this author inPubMed Google Scholar * Randal J. Kaufman View author publications You can also search for this author inPubMed Google
Scholar * Matthias Trost View author publications You can also search for this author inPubMed Google Scholar * Sumana Sanyal View author publications You can also search for this author
inPubMed Google Scholar * Sally A. Cowley View author publications You can also search for this author inPubMed Google Scholar * Pedro Carvalho View author publications You can also search
for this author inPubMed Google Scholar CONTRIBUTIONS M.L.v.d.W. and P.C. designed the study. M.L.v.d.W. performed most of the experiments with the help of N.S. L.J., T.Y.H. and R.J.K.
provided the mouse astrocytes. T.H. prepared the TMT-10-plex samples from mouse astrocytes. M.E.D. and M.T. generated and analysed the mass spectrometry data. S.S. helped in the analysis of
endogenous Tapasin and MHC-I. All work in human iPSCs was supervised S.A.C. and performed by K.S and M.L.v.d.W. M.L.v.d.W. and P.C. analysed the data with the help of all the authors.
M.L.v.d.W. and P.C. wrote the manuscript with input from all the authors. CORRESPONDING AUTHOR Correspondence to Pedro Carvalho. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare
no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review
file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative
Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the
original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in
the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended
use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE van de Weijer, M.L., Samanta, K., Sergejevs, N. _et al._ Tapasin assembly
surveillance by the RNF185/Membralin ubiquitin ligase complex regulates MHC-I surface expression. _Nat Commun_ 15, 8508 (2024). https://doi.org/10.1038/s41467-024-52772-x Download citation *
Received: 21 February 2024 * Accepted: 19 September 2024 * Published: 01 October 2024 * DOI: https://doi.org/10.1038/s41467-024-52772-x SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative
Trending News
Dwp change for people on benefits born within three-year period rolled outThe Department for Work and Pensions is launching a new initiative to support benefits claimants born in specific years....
Ryanair passengers praise 'perfect' cabin bag with 'lots of pockets' - YorkshireLiveWhat's OnRyanair passengers praise 'perfect' cabin bag with 'lots of pockets'The VANKEV Backpack is the ideal travel com...
Moyes in tribute to victims and emergency services after liverpool parade crashTHE EVERTON BOSS BECAME THE LATEST FIGUREHEAD TO EXPRESS SOLIDARITY WITH THOSE AFFECTED BY THE CITY CENTRE INCIDENT 11:3...
Foreign office issues fresh warnings to brits heading to turkeyThe UK Foreign Office has today issued a fresh caution to Brits planning to visit Turkey, advising extra care when using...
Network rail statement over lime street travel delays after paradeHUNDREDS WAITED OUTSIDE LIME STREET TRAIN STATION FOLLOWING THE PARADE AND SUBSEQUENT INCIDENT ON WATER STREET 13:27, 27...
Latests News
Tapasin assembly surveillance by the rnf185/membralin ubiquitin ligase complex regulates mhc-i surface expressionABSTRACT Immune surveillance by cytotoxic T cells eliminates tumor cells and cells infected by intracellular pathogens. ...
Bulls find few places to hide; wal-mart suffers 3% dentBears slapped around the U.S. stock indexes Friday afternoon, driving the Nasdaq under its 50-day moving average line. T...
Reply to: where was wheat domesticated?Access through your institution Buy or subscribe replying to Y. Lev-Mirom and A. Distelfeld _Nature Plants_ https://doi....
High-tech agriculture: farmers risk being ‘locked in’ to unsustainable practicesSince World War II, Europe’s agricultural sector has been very receptive to new technology, and the result has been stag...
Goldman sachs: tiktok generation set to boost these music stocks as streaming surgesGoldman Sachs has said record labels and music publishers are set for a strong rebound as younger audiences continue to ...