Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-t cell bispecific antibody
Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-t cell bispecific antibody"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT T-cell bispecific antibodies (TCBs) crosslink tumor and T-cells to induce tumor cell killing. While TCBs are very potent, on-target off-tumor toxicity remains a challenge when
selecting targets. Here, we describe a protease-activated anti-folate receptor 1 TCB (Prot-FOLR1-TCB) equipped with an anti-idiotypic anti-CD3 mask connected to the anti-CD3 Fab through a
tumor protease-cleavable linker. The potency of this Prot- FOLR1-TCB is recovered following protease-cleavage of the linker releasing the anti-idiotypic anti-CD3 scFv. In vivo, the
Prot-FOLR1-TCB mediates antitumor efficacy comparable to the parental FOLR1-TCB whereas a noncleavable control Prot-FOLR1-TCB is inactive. In contrast, killing of bronchial epithelial and
renal cortical cells with low FOLR1 expression is prevented compared to the parental FOLR1-TCB. The findings are confirmed for mesothelin as alternative tumor antigen. Thus, masking the
anti-CD3 Fab fragment with an anti-idiotypic mask and cleavage of the mask by tumor-specific proteases can be applied to enhance specificity and safety of TCBs. SIMILAR CONTENT BEING VIEWED
BY OTHERS PRECISION-ACTIVATED T-CELL ENGAGERS TARGETING HER2 OR EGFR AND CD3 MITIGATE ON-TARGET, OFF-TUMOR TOXICITY FOR IMMUNOTHERAPY IN SOLID TUMORS Article Open access 30 March 2023 T
CELL-REDIRECTING ANTIBODY FOR TREATMENT OF SOLID TUMORS VIA TARGETING MESOTHELIN Article 10 June 2024 COMPARISON OF ANTIBODY-SCTRAIL FC FUSION PROTEINS WITH VARYING VALENCY FOR EGFR AND
TRAIL RECEPTORS Article Open access 06 May 2025 INTRODUCTION Cancer immunotherapy proves clinical efficacy in several indications1. T-cell bispecific antibodies (TCBs) are antibodies
targeting an antigen expressed on target cells and the CD3ε subunit of the T-cell receptor on T cells to mediate tumor cell lysis. We recently described 2 + 1 TCBs consisting of an inert Fc
region, two tumor antigen-binding Fab fragments and one Fab fragment binding to CD3 on the T-cell receptor2,3. The addition of the Fc part, compared to smaller antibody formats4, increases
the half-life while systemic activation of immune cells via FcγR or complementary binding is prevented by introduction of P329G LALA Fc mutations5. When T- and tumor cells are simultaneously
bound by the TCB, this results in subsequent T-cell activation and potent serial tumor cell killing. Recently, efficacy of a carcinoembryonic antigen (CEA)-specific CEA-TCB (RG7802) was
demonstrated2,3. CEA-TCB efficiently kills tumor cells with high CEA expression while sparing normal cells with low CEA expression. The threshold of T-cell activation is >10,000 CEA
molecules per cell for efficient killing. However, for other TCBs like the folate receptor 1 (FOLR1, FolRα) TCB (Griessinger, #1759) described below such a threshold does not exist, and
related molecules like ImmTacs can kill cells with low target expression in the range of several hundred receptors as recently demonstrated for peptide MHC complexes as target6. Thus,
physiological tissue expression of a given antigen can be critical when developing TCBs or other T-cell activating therapies such as CAR-T cells7. Improving the specificity of TCBs would
increase the number of potential tumor targets. Proteases like serine proteases (e.g. matriptase), cysteine proteases (e.g. cathepsin S) and matrix metalloproteinases (e.g. MMP-2 and MMP-9)
are overexpressed in several cancer types8. Matriptase, matrix metalloproteinase 2 (MMP-2, gelatinase A) and matrix metalloproteinase 9 (MMP-9, gelatinase B) are overexpressed e.g. in
breast- and ovarian carcinoma9,10,11,12,13,14,15,16,17,18,19. MMP-2 and MMP-9 activity was detected in cervical, breast and ovarian carcinoma and ascites of patients with epithelial ovarian
cancer (EOC) but not in the serum of these patients20. While matriptase can be detected in normal epithelial cells, matriptase activity is mainly detected in cancer21. Therefore, these
proteases are suitable as cancer-specific activators of potent agents like TCBs allowing the targeting of otherwise unsuitable antigens. We have previously generated an FOLR1-TCB
(Griessinger, #1759 4, shift the rest). FOLR1 is overexpressed in various tumors including ovarian, lung and breast cancer22, but is also expressed to lower degrees on normal cells e.g. in
the lung and kidney23. While FOLR1-TCB was efficacious in vitro and in xenograft models, severe on-target toxicity in the lung of non-human primates was observed24. Based on this experience,
we chose FOLR1-TCB as a relevant model to show proof-of-concept for masking the anti-CD3 moiety with an anti-idiotypic antibody scFv fused via a protease cleavable linker to the TCB. For
this purpose, we fused a specific anti-idiotypic anti-CD3 scFv N-terminally to the anti-CD3 variable heavy chain connected by a protease cleavable linker and demonstrated that active
proteases located in the tumor microenvironment lead to cleavage and subsequent unmasking of the anti-CD3 targeting moiety. Unmasking results in efficient killing of FOLR1-positive tumor
cells in vitro and in vivo while sparing normal cells with low FOLR1 expression. RESULTS ENGINEERING OF PROTEASE-ACTIVATED ANTIBODIES In non-human primates, on-target toxicity has been
observed when a highly potent FOLR1-TCB (based on clone 16D5) with EC50 values in the single-digit pM range was evaluated in tolerability experiments at single doses as low as 10 µg/kg 24.
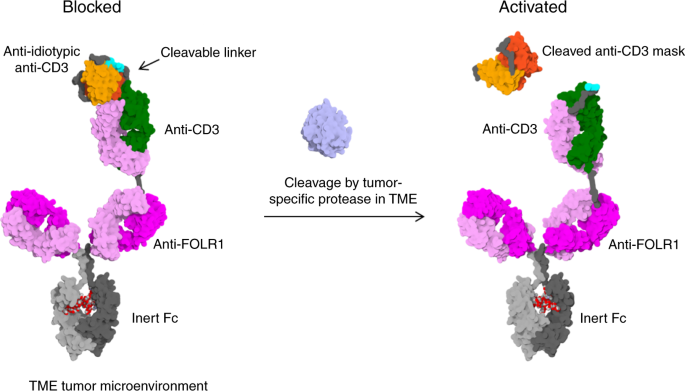
To overcome this limitation, our approach was to block CD3 binding with an anti-idiotypic anti-CD3 scFv that can be cleaved off by tumor-specific proteases. The Prot-FOLR1-TCB is supposed to
be specifically activated in the tumor microenvironment releasing the blocking anti-idiotypic CD3 scFv (Fig. 1). To prove the feasibility of blocking the anti-CD3 Fab, we first engineered a
monovalent anti-CD3 antibody with a N-terminally fused anti-idiotypic anti-CD3 scFv (Fig. 2a). To test the concept of protease-activation in the TCB format, we then engineered a
Prot-FOLR1-TCB by fusion of this anti-idiotypic anti-CD3 scFv to the anti-CD3 Fab (Fig. 2b). The anti-CD3 Fab is a humanized anti-CD3ε Fab with monovalent affinity to human/cyno CD3ε in
single-digit nM range. Anti-idiotypic anti-CD3 antibodies were identified from hybridomas from mice immunized with anti-CD3 F(ab′)2 fragments. Several anti-idiotypic anti-CD3 IgGs were
screened for their avidity to anti-CD3 F(ab′)2 and tested in IgG and TCB format for masking efficiency. From these, the two anti-idiotypic anti-CD3 masks 4.15 and 4.32 were chosen to be
tested in the Prot-FOLR1-TCB format. While the affinity to the anti-CD3 Fab was the same (~2 nM), the affinity to the anti-CD3 Fab in the TCB format was 20 nM for the clone 4.15 and 10 nM
for the clone 4.32 (Supplementary Table 1). The lower affinity of the masking scFv resulted in lower masking-efficiency shown in target cell killing for target cells with very high FOLR1
expression (Supplementary Fig. 1A, B). As both masks were released resulting in comparable potency of activated Prot-FOLR1-TCB, we chose the 4.32 mask for further characterization
(Supplementary Fig. 1A, B). The Prot-FOLR1-TCB was generated by fusing the anti-idiotypic anti-CD3 scFv to the variable heavy (VH) chain of the anti-CD3 Fab fragment (Fig. 2b, c). An
additional disulfide bridge (VH44-VL100) was inserted to increase stability and reduce aggregate formation of IgG-scFv fusion molecules24,25,26,27. The variable heavy and the variable light
chain of the scFvs were connected by a (G4S)4 linker. For the Prot-FOLR1-TCB (clone 16D5), a common light chain for both the FOLR1 and anti-CD3 Fab fragment was applied, facilitating correct
light chain association and “knobs-into-hole” technology to enable correct Fc heterodimerization28,29. All antibodies carried an inert Fc with P329G LALA5 mutations. Fusion of the
anti-idiotypic anti-CD3 scFv was achieved via a synthetic linker sequence (33 amino acids) comprising different protease cleavage sites (Supplementary Table 2). FOLR1-TCB without a blocking
moiety or a respective Prot-FOLR1-TCB with a noncleavable linker formed by a (G4S)3−(G7S)1−(G4S)2 linker were used as controls (Supplementary Table 2). An analogous protease-activated
mesothelin TCB (Prot-MSLN-TCB) was generated using a CrossMAbVH-VL format29,30 with charged residues31 in the Fab fragments to enable correct light chain pairing based on the humanized
Mesothelin antibody SS1 (Fig. 2c). All antibodies were transiently produced in HEK293 cells, purified and analyzed for integrity and monomer content (Supplementary Table 3). The antibodies
were stable once produced and the linkers, containing different cleavage sites, were stable during the purification process. The protease-activated IgGs and TCBs containing either a
matriptase (matA site) or a MMP2, -9-matriptase (MMP-matA site) cleavage site were additionally analyzed for stability and cleavage with recombinant human matriptase by capillary
electrophoresis. The antibodies were stable at 4 °C and at 37 °C for a minimum of 48 h and were cleaved in vitro by recombinant human matriptase (Supplementary Fig. 2). In order to
demonstrate the cleavage site specificity, the constructs were incubated either with recombinant human matriptase (rhMatriptase), recombinant human MMP-2 (rhMMP-2) or recombinant human MMP-9
(rhMMP-9). The cleavage of the linker was indirectly measured in a cell assay via luminescence, indicating activation of Jurkat NFAT-cells upon CD3 binding. The MMP site used herein was
cleaved by rhMMP-2 and rhMMP-9, whereas the matA cleavage site was cleaved by rhMatriptase (Supplementary Fig. 3). In order to further demonstrate the thermal stability of the protease
linkers, different antibodies, containing an MMP or a combined MMP-matA cleavage site, were heated up to 85 °C and analyzed for aggregate formation and melting temperature. All antibodies
tested were stable up to 58 °C (Supplementary Table 4). Additionally, no cleavage was detectable for the Prot-FOLR1-TCB containing matA or MMP-matA site after incubation in human serum for
14 days at 37 °C (Supplementary Fig. 4). PROT-MΑCD3 IGG CAN BIND TO CD3Ε ANTIGEN AFTER ACTIVATION To further characterize the antibodies and prove feasibility of re-activation of the
anti-CD3 Fab fragment, we verified the globular integrity and the functional activity of the protease-activated monovalent anti-CD3 IgG (Prot-mαCD3 IgG) by negative stain transmission
electron microscopy (NS-TEM) and atomic force microscopy (AFM). Class-averages derived from multivariate statistical analysis of particles in micrographs recorded with NS-TEM confirmed the
expected globular structure of the Prot-mαCD3 IgG (Fig. 3a). The resolution achieved with this method allowed to distinguish between three globular fragments. The C-terminal domain of the
triangularly shaped Fc-fragment was linked to an anti-CD3 Fab fragment which was characterized by a central hole. The distal N-terminal domain resembled the anti-idiotypic anti-CD3 scFv
masking moiety and had a size expected for VH/VL domains, whereas molecules lacking this moiety were shorter. Molecule complexed with CD3-Fc antigen fusion were larger and characterized by
two Fc fragments at the distal ends. Unfortunately, the achieved given resolution was not sufficient to annotate them (Fig. 3a). The unmasking functionality of the construct was structurally
confirmed on the level of single molecules using tapping-mode AFM in liquid. Masked Prot-mαCD3 molecules were trapped on a surface of mica and monitored for structural changes introduced by
matriptase treatment and the addition of the Fc-CD3εδ ligand. Topology and size changes were observed and attributed to the unmasking-complexation process (Fig. 3b and Supplementary Fig.
5). The changes match the molecule structures observed with NS-TEM with respect to topology and length,. Thus, at the beginning of the experiment, the molecules resembled a chain of three
jointed segments. The treatment with matriptase resulted in a fraction of shorter molecules with only two segments, and the treatment with the more bulky Fc-CD3εδ ligand finally resulted in
the expected elongated complex over time. MASKING OF ANTI-CD3 FAB IMPAIRS CD3 BINDING ON T CELLS To prove that crosslinking of the monovalent anti-CD3 binder is required for successful
T-cell activation, we compared the mαCD3 IgG both in the presence or absence of plate-coated anti-human Fc antibody, using Jurkat NFAT-cells or peripheral blood mononuclear cells (PBMCs) as
effector cells (Fig. 4). The Jurkat NFAT reporter cell line contained a nuclear factor of activated T-cell (NFAT) promoter upstream of a luciferase gene. Binding and subsequent crosslinking
of CD3ε induces downstream signaling resulting in luciferase expression which was quantified via luminescence after substrate addition. In addition, activation of natural T cells in the PBMC
fraction was analyzed by FACS using the T-cell activation marker CD69. We clearly detected dose-dependent Jurkat NFAT and CD8 T-cell activation only in the presence of plate-coated
anti-human Fc antibody, indicating that this activation was dependent on crosslinking of the monovalent CD3 antibody. In contrast, no activation was detected in the absence of anti-human Fc
antibody coated on plates (Fig. 4a, b). In subsequent assays we used the Jurkat NFAT reporter assay or human PBMCs to analyze CD3 binding of Prot-mαCD3 IgG (masked, cleaved or noncleavable)
compared to mαCD3 IgG. The Prot-mαCD3 IgG, containing an matA site, was activated by cleavage with rhMatriptase to show full activity (Fig. 4c, d). In addition, blocking of CD3 binding was
clearly dependent on the masking by anti-idiotypic CD3 scFvs as the N-terminal fusion of an unrelated Fab did not block CD3 binding (Fig. 4c, d). MASKING-EFFICIENCY DEPENDS ON ANTIGEN
EXPRESSION LEVEL In order to demonstrate the reduction of target cell lysis by blocking of the anti-CD3 Fab, the dose-dependent T-cell killing of FOLR1-positive tumor cells by
Prot-FOLR1-TCBs was measured after 48 h. Masking-efficiency was investigated for HeLa and Skov-3 cells that express high (approx. 2 mio antigen-binding sites (ABS)/cell) or medium (approx.
0.1 mio ABS/cell) levels of FOLR1 antigen, respectively. Prot-FOLR1-TCB, precleaved with rhMatriptase, showed dose-dependent killing for both cell lines (Fig. 5a). The Prot-FOLR1-TCB
containing a noncleavable site did not induce any killing for Skov-3 cells in the indicated concentration range. For HeLa cells with a very high FOLR1 expression, the EC50 of the
Prot-FOLR1-TCB (noncleavable site) was significantly reduced up to 4000-fold compared to the cleaved TCB (Fig. 5a). The activity of Prot-FOLR1-TCB was restored after linker cleavage. Several
proteases are described to be overexpressed in ovarian carcinoma. We chose matrix metalloproteinase-2,-9 (MMP-2, MMP-9) and matriptase to compare cleavage by cellular proteases. We compared
target cell cytotoxicity for Prot-FOLR1-TCB containing either an MMP site, a matriptase (matA) site or a combined MMP-matA site for cleavage by naturally expressed proteases (Supplementary
Fig. 6A, B). Dose-dependent killing assays using FOLR1-positive HeLa and Skov-3 cells revealed a higher potency for the Prot-FOLR1-TCB with the combined MMP-matA site (EC50 approx. 6- to
7-fold lower for HeLa cells, EC50 approx. 2−3-fold lower for Skov-3 cells) (Supplementary Fig. 6A, B). For this reason the MMP-matA site was chosen for further evaluation. To analyze if the
potency of the Prot-FOLR1-TCB can be recovered after linker cleavage, we compared FOLR1-TCB, Prot-FOLR1-TCB (precleaved with rhMatriptase) and Prot-FOLR1-TCB for dose-dependent target cell
cytotoxicity and Granzyme B release using HeLa and Skov-3 cells (Fig. 5c–f). The Prot-FOLR1-TCB was cleaved by cellular proteases and its potency was comparable to the precleaved
Prot-FOLR1-TCB and the FOLR1-TCB using HeLa cells with high FOLR1 expression. For Skov-3 cells with medium FOLR1 expression the potency of the Prot-FOLR1-TCB was lower compared to the
precleaved Prot-FOLR1-TCB and the FOLR1-TCB (Fig. 5c–f). However, the potency of the fully activated (precleaved) Prot-FOLR1-TCB was comparable to FOLR1-TCB for both cell lines (Fig. 5c–g).
The Prot-FOLR1-TCB with a glycine and serine (GS) noncleavable site induced significantly less T-cell-mediated cytotoxicity for HeLa and Skov-3 cells compared to the Prot-FOLR1-TCB
containing an MMP-matA site. At the highest concentration (10 nM), a maximal T-cell-mediated cytotoxicity of 20% was observed for highly FOLR1-positive HeLa cells (Fig. 5c). Quantification
of cytotoxic granule granzyme B after incubation of target cells with PBMCs and Prot-FOLR1-TCB revealed a dose-dependent TCB-mediated release of granzyme B. The granzyme B release mediated
by the activated Prot-FOLR1-TCB was comparable to the FOLR1-TCB whereas no granzyme B release could be detected for the masked Prot-FOLR1-TCB with a noncleavable linker (Fig. 5e, f). In
order to analyze the kinetics of T-cell-mediated cytotoxicity mediated by the Prot-FOLR1-TCB, we investigated tumor cell growth for MDA-MB-231 NucLight red cancer cells (medium FOLR1
expression) during coincubation with PBMCs and the Prot-FOLR1-TCB. We detected significant growth inhibition for the Prot-FOLR1-TCB with the MMP-matA site compared to the Prot-FOLR1-TCB
containing a noncleavable site (Fig. 5g). THE CONCEPT IS APPLICABLE FOR OTHER TCBS A major advantage of masking the anti-CD3 Fab fragment is that the concept can be applied for TCBs
targeting different tumor antigens. For proof-of-concept we analyzed dose-dependent T-cell-mediated cytotoxicity on mesothelin (MSLN)-positive tumor cell lines NCIH596 (approx. 80,000 MSLN
ABS/cell) and AsPC1 (approx. 50,000 MSLN ABS/cell) mediated by an analogous Prot-MSLN-TCB. The precleaved Prot-MSLN-TCB and the MSLN-TCB were comparable regarding their potency for both cell
lines (Supplementary Fig. 6c, d). The Prot-MSLN-TCB, cleaved by proteases expressed only by the target cell line, was comparable to the MSLN-TCB for NCI H596 cells (Supplementary Fig. 6C),
whereas for AsPC-1 cells (Supplementary Fig. 6D), the in vitro activated Prot-MSLN-TCB did not reach the potency of the MSLN-TCB. The masked MSLN-TCB containing a GS noncleavabe linker
prevented T-cell-mediated killing in the indicated concentration range for both cell lines. CYTOTOXICITY OF FOLR1-TCB IS ABOLISHED BY BLOCKING ANTI-CD3 As FOLR1 is known to be expressed to a
low extent by normal lung and kidney cells23, we tested the reduction of potential on-target toxicity by analyzing target cell killing of Prot-FOLR1-TCB on primary human bronchial
epithelial cells (HBEpiC, <1000 FOLR1 ABS/cell) and primary human renal cortical epithelial cells (HrcEpiC, <1000 FOLR1 ABS/cell). Neither killing nor T-cell activation (CD69 increase
for CD8 T cells) was observed for HBEpiC and HrcEpiC cells using Prot-FOLR1-TCB and huPBMCs (Fig. 6a–d). In contrast, the parental FOLR1-TCB induced cell lysis and T-cell activation for both
cell types (Fig. 6a–d). PROT-FOLR1-TCB CAN BE ACTIVATED BY CANCER EXPLANTS In order to show that the Prot-FOLR1-TCB can be activated by human patient-derived samples expressing FOLR1 as a
tumor target antigen, we set up a method to analyze tumor samples without digestion to exclude artefacts coming from tumor digestion. This might be of importance, as not only tumor cells but
also cells from the tumor microenvironment (e.g. tumor-associated macrophages (TAM)) are described to express MMP-2 and MMP-932. Protease-activated TCBs containing linkers with different
protease sites were incubated with mechanically cut tumor pieces before CD3-mediated T-cell activation was analyzed. In this setting, binding of the cleaved TCB to tumor cells and Jurkat NFA
cells results in induction of a luciferase signal. Jurkat NFAT activation was detected for a benign FOLR1-positive ovary sample using the FOLR1-TCB but no activation was detected using the
Prot-FOLR1-TCB (Fig. 7a). However Jurkat NFAT reporter cells were activated after incubation of FOLR1-positive ovarian tumor samples with FOLR1-TCB or Prot-FOLR1-TCB containing an MMP-matA
site (Fig. 7b, c). PROT-FOLR1-TCB IS EFFICACIOUS IN VIVO As stability of Prot-FOLR1-TCB in human serum over time was shown (Supplementary Fig. 4), the stability of Prot-FOLR1-TCB was
analyzed in vivo. Bioavailability of active Prot-FOLR1-TCB 7 days after intravenous injection revealed that the Prot-FOLR1-TCB containing the combined MMP-matA site was cleaved to some
extent (bioavailability around 35%) in non-tumor-bearing mice. Furthermore, we determined the serum bioavailability of the Prot-FOLR1-TCBs containing the single MMP site or the single matA
site. Both were also cleaved to some extent (MMP ~25%, matA ~14%), however, less than the combined linker. In this analysis we included also a Prot-FOLR1-TCB with a new matriptase (matB)
cleavage site and a matC site, described to be cleaved by matriptase33 (Supplementary Table 2). Both Prot-FOLR1-TCBs had a very low serum bioavailability of ~5% in non-tumor-bearing mice
(Fig. 8a). Thus, we used these Prot-FOLR1-TCBs in a subsequent efficacy study using a breast PDX (patient-derived xenograft) “BC004” model. Immunohistochemistry staining confirmed FOLR1 and
matriptase expression in this PDX BC004 model (Fig. 8b). Tumor growth inhibition (TGI) was evaluated in an efficacy study in stem cell humanized NSG mice with autologous T cells and the
respective TCBs at a dose of 4 mg/kg. FOLR1-TCB, Prot-FOLR1-TCB containing the matB site and the matC site showed significant TGI at day 62 compared to the vehicle group (Fig. 9a). The
Prot-FOLR1-TCB containing a noncleavable (non-cleav) site was not different from vehicle showing that masking prevented antitumor efficacy. Comparing Prot-FOLR1-TCB with the matB site and
the matC site, the latter one was more efficacious and comparable to the parental FOLR1-TCB (Fig. 9a). Immuno-pharmacodynamics was analyzed by quantification of human CD3 T cells in the
tumor. All treatment groups were significantly different from the vehicle group (Fig. 9b), whereas the FOLR1-TCB and the Prot-FOLR1-TCB containing matB site were not significantly different.
Prot-FOLR1-TCB containing matB site was also significantly different from Prot-FOLR1-TCB with noncleavable site, whereas the Prot-FOLR1-TCBs with matC site was not different from
Prot-FOLR1-TCB with noncleavable site (Fig. 9b). Comparison of serum bioavailability of active Prot-FOLR1-TCB in non-tumor-bearing humanized mice and in tumor-bearing mice showed no evidence
for tumor-leakage of activated Prot-FOLR1-TCB into the serum as bioavailability of active Prot-FOLR1-TCB was comparable in non-tumor- vs. tumor-bearing mice (Fig. 9c, d). DISCUSSION CD3
targeting antibodies can be used for different purposes: the muromonab-CD3 antibody was approved to prevent allograft rejection after organ transplantation34,35. However, systemic T-cell
activation led to side effects with strong cytokine release. Humanization and prevention of FcR binding improved the safety profile of anti-CD3 antibodies for autoimmune diseases but
systemic T-cell activation remains a challenge. T-cell bispecific antibodies are promising agents to mediate potent tumor cell killing but they require a tumor antigen with expression
restricted to tumor cells to avoid on-target/off-tumor toxicity on normal cells24. The development of TCBs for cancer immunotherapy may therefore profit from a protease-activated anti-CD3
moiety to reduce systemic side effects. Here we describe a novel protease-activated 2 + 1 Prot-FOLR1-TCB with a masked anti-CD3 Fab fragment and an inert Fc-region. T-cell activation
requires binding of FOLR1-TCB to both FOLR1 and CD3. To avoid this in the periphery, the anti-CD3 Fab is blocked until activation by linker cleavage through a tumor-specific protease site
(matriptase, MMP-2 or MMP-9) that connects the mask and anti-CD3 Fab. FOLR1 is overexpressed in ovarian23,36 or lung cancer23 but also expressed in normal tissue (like lung and kidney23)
making it a suitable target for proof-of-concept. FOLR1 targeting antibodies e.g. farletuzumab37 and antibody–drug conjugates e.g. IMGN853 appear to be safe in in clinical trials37,38,39.
However, targeting FOLR1 with TCBs resulted in on-target/off-tumor toxicity as FOLR1-TCBs can induce killing of normal cells with few hundred FOLR1 receptors. In line with this, toxicity was
observed in non-human primates after injection of 10 µg/kg FOLR1-TCB24 and in patients with ovarian cancer treated with the first-generation bispecific antibody OC/TR F(ab′)2 targeting
FOLR1 and CD340. Nevertheless, FOLR1-TCBs may be advantageous compared to FOLR1 antibodies due to their higher antitumor potency and NK-cell-independent mechanism41. The concept of
tumor-specific protease-activation has been described previously (reviewed in refs. 42,43,44). Active proteases located in the tumor microenvironment lead to activation of tumor targeting
moieties by cleavage of the linker and consecutive unmasking of the targeting moiety. Proof-of-concept was shown for tumor-specific protease-activation of a prodrug that can reduce on-target
toxicity for an EGFR-targeted antibody with N-terminally fused blocking peptides and a protease cleavable linker33,45. Other examples are an anti-PD-L1 probody CD-71 targeting probody drug
conjugate46,47. Similarly, this approach was described for a protease-activated CTLA-4 antibody48 and using a coiled-coil masking domain for CD20, HER2 and CD3 antibodies49 This strategy of
protease-activation demonstrated enhanced tolerability in (pre-)clinical studies while retaining antitumoral efficacy. Here, we chose not to block the tumor targeting moiety, but rather the
anti-CD3 targeting moiety in a TCB in order to generate a platform applicable to various tumor antigens. Contrary to peptide blocking moieties the anti-idiotypic anti-CD3 scFvs, described
here, can be humanized decreasing the risk of immunogenicity50,51. Our results show that it is feasible to block CD3 binding of anti-CD3 Fab fragments in IgG-like antibodies by an
anti-idiotypic disulfide-stabilized anti-CD3 scFv and to unblock this by tumor-specific proteases52. First, we generated a monovalent anti-CD3 IgG with one anti-idiotypic anti-CD3 scFv with
an affinity of approx. 2 nM. When the scFv was attached to both anti-CD3 Fab fragments in a bivalent homodimeric IgG (Supplementary Fig. 7), the corresponding construct could not be properly
purified due to its high aggregation tendency following pH neutralization after affinity chromatography, which is likely a consequence of intramolecular aggregation of scFvs. The globular
structure of Prot-mαCD3 IgG and the validity of the matriptase-triggered de-masking concept is substantiated by single particle analytics. NS-TEM data of complexes made from CD3 antigen and
activated Prot-mαCD3 IgG confirm that the distal region is crucial for complex formation. Matriptase-driven unmasking demonstrated individual molecule level, in situ, with tapping-mode AFM.
In a second step blocking of the anti-CD3 moiety by the anti-idiotypic, anti-CD3 scFv could be also shown in the TCB format. Target cell cytotoxicity was significantly reduced for the
Prot-FOLR1-TCB containing a noncleavable site. Notably, the masking efficiency, in the Prot-TCB format correlated with the antigen expression level of the target cells and with the affinity
of the anti-idiotypic anti-CD3 binder to the anti-CD3 in the TCB. Different proteases are described to be active in several cancers like serine and cysteine proteases as well as matrix
metalloproteinases8,53,54,55. Typically, the expression and the activity of these proteases is minimal in normal tissue, so that these proteases can be exploited for tumor-specific
activation or imaging. For proof-of-concept we have focused on MMP-2, -9 and matriptase for the Prot-FOLR1-TCB as these proteases are known to be overexpressed and active in ovarian
carcinoma9,10,12,56 with minimal activity in normal tissue20,33,53. LeBeau et al.21 showed that matriptase is expressed in normal colon, but the active form of matriptase was not detected
there. Demeter et al.20 showed that both MMP-2 and MMP-9 are not active in the serum of patients but are active in the ascites and tumors of recurrent patients with EOC. To prove the
stability of our protease-activated antibodies, we showed the integrity after incubation in human serum for 14 days at 37 °C. Two different matriptase cleavage sites were introduced into the
generated constructs. For the construct containing the RQRRVVGG matriptase cleavage site, we observed that it was cleaved during production in HEK293 cells which was attributed to a furin
cleavage site within this linker. Comparing Prot-FOLR1-TCB using different linkers solely cleaved by cellular proteases, we confirmed a synergistic effect for the combination of the cleavage
sites for MMP-2, -9-matriptase (MMP-matA) compared to matriptase (matA) or MMP-2, -9 (MMP) linkers alone. Several linkers described for protease-activated antibodies currently under
investigation also contain 2−3 substrate sequences for different proteases. One example is an FAP-CD95L fusion protein containing an MMP-2/uPA cleavable linker57. Notably, the potency of the
Prot-FOLR1-TCB containing an MMP-2, -9-matriptase linker was comparable to FOLR1-TCB using HeLa or Skov-3 cells as target cells. Target cell lysis correlated with T-cell activation and
secretion of granzyme B induced by the crosslinking through FOLR1-positive cancer cells as it was recently shown for CEA TCB2. For primary human bronchial epithelial cells and primary human
renal cortical cells expressing low amounts of FOLR1, the masking significantly reduced target cell killing even at concentrations tenfold higher than the one used for tumor cells. Reduced
on-target toxicity was also shown for the EGFR-targeting Probody described by Desnoyers et al.33 in non-human primates compared to cetuximab. Similarly, Watermann et al. observed reduced
liver-toxicity mediated by the protease-activated FAP-CD95L fusion protein compared to the parental molecule. These data provide evidence that cleavable masks can enhance safety of compounds
targeting antigens whose expression is not restricted to the tumor cells. Importantly, the potency of the Prot-FOLR1-TCB was fully recovered after cleavage with recombinant human
matriptase. Additionally, we showed activity of the Prot-FOLR1-TCB containing MMP-2, -9-matriptase cleavable linker in undigested human ovarian tumor samples. For this purpose we applied
undigested tumor samples as it was reported that cells of the tumor microenvironment are involved in protease expression e.g. TAM or fibroblasts58,59,60,61. In order to check for FOLR1 and
protease expression in this assay without the need to monitor pre-existing T cells and interference from debris and dead cells common in human tumor explants, we developed an assay based on
Jurkat cells expressing luciferase under the control of an NFAT-inducible reporter. Activation of the Prot-FOLR1-TCB containing an MMP-matA site was observed for two undigested FOLR1 +
ovarian carcinoma samples whereas no activation occured in a benign FOLR1 + ovarian sample. This is in line with reports describing higher MMP-2 and MMP-9 activity in advanced EOC or
metastasis compared to benign tumors18,20 and their role in extracellular matrix degradation and activation of growth factors to facilitate invasion and tumor growth32,55,62. To demonstrate
the applicability of the protease-activated anti-CD3 Fab fragment for other targets, we engineered an analoguous protease-activated mesothelin TCB (Prot-MSLN-TCB). We confirmed efficient
masking of the Prot-MSLN-TCB with a noncleavable linker and comparable efficacy of the activated Prot-MSLN-TCB and the MSLN-TCB using MSLN-positive target cells. Finally, aiming to analyze
the Prot-FOLR1-TCB containing MMP-matA site in vivo, we first tested the stability in non-tumor-bearing mice. We observed that the Prot-FOLR1-TCBs with MMP, matA and MMP-matA sites were
cleaved to some extent in the absence of tumor in vivo. The Prot-FOLR1-TCBs containing the single cleavage sites were cleaved to a lower extent (MMP ~25%, matA ~14%) while the combination of
both cleavage sites resulted in enhanced cleavage (combined MMP-matA ~35%). This finding was unexpected as all linkers were stable in human serum. In order to find a suitable cleavage site,
we analyzed the in vivo stability of Prot-FOLR1-TCB containing the matriptase site matB (PMAKK) or the matriptase site matC (LSGRSDNH) which was described to be stable in cynomolgous
monkeys as a positive control33. Both Prot-FOLR1-TCBs containing matB or the matC site had low serum bioavailability of ~5% in non-tumor-bearing mice and thus were evaluated for their
antitumor activity in vivo. Tumor growth inhibition was evaluated in an efficacy study using an orthotopic breast PDX BC004 model in humanized mice with autologous T cells. Prot-FOLR1-TCB
containing a noncleavable linker behaved comparable to vehicle regarding TGI whereas the FOLR1-TCB, Prot-FOLR1-TCB containing the matB site and the matC site were significantly different
from vehicle. The Prot-FOLR1-TCB containing the matB site induced superior TGI from all Prot-FOLR1-TCBs, suggesting that the matriptase cleavage of this site may be more efficient than the
matriptase cleavage of matC. Importantly, the comparison of bioavailabilities of active Prot-FOLR1-TCB in non-tumor-bearing and tumor-bearing mice did not suggest any tumor leakage of
Prot-FOLR1-TCB from the tumor into the serum. As an additional safety measure for targets that are expressed to a low extent on normal cells, this novel protease-activated anti-CD3 moiety
could be used to engineer TCBs for improved specificity and therefore increase the number of targets amenable for TCBs. METHODS CONSTRUCTION OF PROTEASE-ACTIVATED ANTIBODIES The variable
chains of the scFvs were connected by a (G4S)4 linker and cysteins were inserted (VH44-VL100) for disulfide stabilization25,26. Single chain variable fragment (scFv) sequence synthesis was
ordered at Invitrogen, including the necessary restriction sites for cloning. Single chain Fv DNA sequences of three anti-idiotypic anti-CD3 antibodies were N-terminally fused in frame with
the anti-CD3 Fab-Fc chain preinserted into the respective recipient mammalian expression vector. The construction of expression vectors for TCBs was performed according to standard
recombinant DNA technologies. All antibody chain genes were separately inserted into expression vectors under the control of the MPSV or CMV promoter (myeloproliferative sarcoma virus or
cytomegalovirus) and transiently expressed in HEK293 cells. The anti-idiotypic single chain fragments (scFv) were fused to the anti-CD3 variable heavy chain (VH) in the respective chain of
TCB. In order to get high yields of correctly paired molecules, the “knobs-into-holes” (KiH) technology was used for heterodimerization28. P329G, L234A and L235A (PG LALA) mutations were
inserted in CH3 and CH2 to prevent binding to FcγRs and C1q63. For cases when no common light chain could be used (MSLN-TCB and Prot-MSLN-TCB), a CrossMAbVH-VL format29,32 and charged
residues in constant chains31 were used to assure correct light chain pairing. CELL LINES HEK293, HeLa and Skov-3 cells were purchased from ATCC (American Type Culture Collection (ATCC)),
MDA-MB-231 NLR were purchased from Essen Bioscience (Cat.# 4487), human bronchial epithelial cells (HBEpiC, 3210) and human renal cortical epithelial cells (HrcEpiC, 4110) were purchased
from ScienCell Research Laboratories, AsPC-1 (ECACC, 96020930) cells were obtained from the European Collection of Cell Cultures (ECACC) and NCI H596 cells were provided by Roche Innovation
Center Munich. Jurkat NFAT-cells were purchased from Promega. All cells were routinely cultured at 37 °C and 5% CO2 and tested for mycoplasma contamination. The cell identity of all tumor
cell lines was verified by FTA cell authentication service provided by the ATCC64. Antigen-binding sites were determined using QIFIKIT® (Dako) according to the manufacturer’s instructions.
KILLING ASSAYS Human peripheral blood mononuclear cells (PBMCs) were purified from buffy coats of healthy donors, obtained from Blutspende Zürich SRK, by conventional histopaque gradient
(Sigma-Aldrich). Blutspende Zürich SRK confirmed that all donors consented into the use of the sample for research purpose. Adherent target cells were trypsinized (0.05 % trypsin/EDTA;
Gibco) and counted using a Vi-CELL device (Beckman Coulter). 20,000 target cells per well were seeded in flat-bottom 96-well plates (tissue culture test plates from TPP) and incubated for
approx. 20 h at 37 °C, 5% CO2 before antibodies and human PBMC effector cells were added (E:T ratio of 10:1). Target cell killing was measured after 48/72 h of incubation at 37 °C, 5% CO2 by
quantification of lactate dehydrogenase (LDH) release into cell supernatants by dead cells (LDH detection kit; Roche Applied Science). Minimal lysis refers to target cells incubated with
effector cells without any TCB. T-cell activation was analyzed by quantification of CD69 for CD8-positive T cells using a MACSQuant device (Miltenyi Biotec)65. Cytokine (IFN-γ, TNF-α, IL-2,
GM-CSF) and cytotoxic granule (granzyme B) secretion was assessed 48 h after incubation of target cells with TCB and PBMCs (as above) using Human Soluble Protein Master Buffer Kit (BD
Biosciences) according to the manufacturer’s protocol. The cleavage of the Prot-FOLR1-TCB was done by incubation of 1 μl of purified recombinant human matriptase (0.44 mg/ml, R&D
Systems) with approx. 10 nmol of antibody in histidine buffer (20 mmol/l, pH 6, Bichsel) for 24 h at 37 °C. The precleaved TCB was not purified after incubation. KINETIC OF TUMOR CELL GROWTH
USING INCUCYTE Tumor cell growth inhibition mediated by Prot-FOLR1-TCB was assessed on CellPlayer™ MDA-MB-231 NucLight Red cells (Essen BioScience) naturally expressing FOLR1. Adherent
target cells were trypsinized (0.05% trypsin/EDTA; Gibco) and 5000 cells per well were seeded in flat-bottom 96-well plates (tissue culture test plates from TPP) before molecules and human
PBMC effector cells were added (E:T ratio of 10:1). All samples were performed in triplicates and incubated in IncuCyte (Essen BioScience) at 37 °C, 5% CO2. Tumor cell growth was analyzed by
target-cell count. The first scan that was carried out approx. 2 h after the addition of PBMCs and TCBs is indicated as time 0 h. The cleavage of the Prot-FOLR1-TCB was done by incubation
of 1 μl of purified recombinant human matriptase (0.44 mg/ml, R&D Systems) with approx. 10 nmol of antibody in histidine buffer (20 mmol/l, pH 6, Bichsel) for 24 h at 37 °C. The
precleaved TCB was not purified after incubation. REPORTER ASSAY USING PATIENT-DERIVED CANCER EXPLANTS We used a Jurkat-NFAT reporter cell line (Promega) to check target expression (FOLR1)
and protease activity in patient-derived undigested human tumor samples. Tumor samples (Indivumed GmbH, Germany) were shipped overnight in transport medium. Approximately, 24 h after
surgery, the sample was cut into small pieces (<1 mm in diameter) before 2−3 pieces were placed into wells before 50 nM of TCBs was added. In one experiment (Fig. 7a), the pieces were put
in 24-well plates prepared with Millicell Cell Culture Insert, 12 mm, hydrophilic PTFE, 0.4 µm (PICM01250, MerckMillipore); in the other experiment (Fig. 7b, c) the 2−3 pieces were put in
wells of a 96-well plate prepared with matrigel (Corning/VWR). Pieces were covered with Matrigel and hardened for 2 min at 37 °C. 50 nM of TCBs was incubated with tumor pieces for 48 h at 37
°C, 5% CO2. Jurkat-NFAT reporter cells were harvested and viability was assessed using ViCell. 500,000 Jurkat NFAT-cells/well were added for 24-well plate and 50,000 Jurkat NFAT-cells/well
were added for 96-well plate. The plates were incubated for 5 h at 37 °C in a humidified incubator before ONE-Glo substrate solution (Promega) was added to each well and incubated for 10 min
at room temperature in the dark. Luminescence was detected using WALLAC Victor3 ELISA reader (PerkinElmer2030), 1 s/well as detection time. MOUSE MODEL All mice were maintained under
specific pathogen-free condition with daily cycles of 12 h light/12 h darkness. The animal facility has been accredited by the Association for Assessment and Accreditation of Laboratory
Animal Care (AAALAC). All animal studies were performed in accordance with the Federation for Laboratory Animal Science Associations (FELASA). The animal studies were approved by and done
under license from the Government of Upper Bavaria (Regierung von Oberbayern; Approval number: Az 55.2.1.54-2532.0-10-16). Animals were maintained for 1 week after arrival to get accustomed
to the new environment and for observation. Daily continuous health monitoring and weekly body weight measurement was conducted. Hematopoietic stem cell humanized mice (humanized mice), used
for efficacy or single-dose PK studies, were generated in-house. Briefly, 4–5-week-old female NOD scid gamma (NSG) mice (Jackson Laboratory, Sacramento, CA USA) were injected i.p. with 15
mg/kg Busulfan (Busilvex, Pierre Fabre Limited) in a total volume of 200 μl. Twenty-four hours later, mice were injected intravenously (i.v.) with 1 × 105 CD34+ cord blood cells (STEMCELL
Technologies Inc, Grenoble, France). Fifteen weeks after cell injection, mice were bled and screened for successful humanization by flow cytometry. In some single-dose PK studies,
8−10-week-old female NOD scid gamma (NSG) mice (Jackson Laboratory, Sacramento, CA USA) were used without any humanization. SINGLE-DOSE PK AND STABILITY STUDY NSG or non-tumor-bearing and
tumor-bearing humanized NSG mice received a single injection of different antibodies (equimolar doses). Seven days post infusion mice were bled under anesthesia (retro-orbital). Fresh blood
was collected in serum separator tubes (Sarstedt, Nuembrecht, Germany) and after centrifugation, serum was frozen and stored at −20 °C for further analysis. PATIENT-DERIVED XENOGRAFT (PDX)
MODEL The human breast cancer patient-derived xenograft HER2+ ER− xenograft model BC_004 was purchased from OncoTest (Freiburg, Germany). Tumor fragments were digested with Collagenase D and
DNase I (Roche), counted and 2 × 106 BC004 cells were injected in total volume of 20 µl PBS into the mammary fat pad. Treatment was started once tumors reached an average volume of
approximately 200 mm3. THERAPEUTIC ANTIBODY TREATMENT A total of nine animals were assigned per group. No statistical methods were used to predetermine the total number of animals needed for
this study; however, taking into consideration the heterogeneity of tumors growth as well the heterogeneous humanization rate of NSG mice, we experienced nine mice per group as a good
number for statistical power. All mice were injected i.v. with 200 µl of the appropriate solution. The mice in the vehicle group were injected i.v. with Histidine buffer (20 mM Histidine,
140 mM NaCl, pH 6.0) and the treatment group with the antibody diluted with Histidine buffer to a volume of 200 µl. Mice received once weekly injections of 4 mg/kg of each compound
(equimolar doses, so 3.6 mg/kg of TCB) and a total of four treatments. TUMOR VOLUME MEASUREMENT Tumor volume (½ [length × width2]) was measured three times per week by caliper. NECROPSY AND
IMMUNOHISTOCHEMISTRY At study termination, mice were sacrificed and tumors were surgically removed from all animals. Some tumors were harvested at start of treatment for the baseline
characterization by immunohistochemistry. All tissue samples were fixed in 10% formalin (Sigma, Germany) and processed for FFPET (Leica 1020, Germany). Four-micrometer paraffin sections were
subsequently cut in a microtome (Leica RM2235, Germany). Human Matriptase immunohistochemistry was performed with anti-human ST-14 (PA5-29764 from Thermo Scientific, Germany), human folate
receptor alpha with anti-FOLR1 (BN3.2, Byosystems, Switzerland) and human T-cell detection with anti-CD3 (ab5690, Abcam, Germany). Stainings were performed in the Leica autostainer (Leica
ST5010, Germany) following the manufacturer’s protocols. Sections were counterstained with hematoxylin (Sigma-Aldrich) and slides were scanned using Olympus VS120-L100 Virtual Slide
Microscope scanner. Quantification of human CD3-positive cells from scan images was performed with Definiens software (Definiens, Germany). For this, whole scans were uploaded in the tissue
developer module and necrotic areas were excluded with segmentation analysis. Secondly, a threshold was set to recognize the brown staining of the targeted CD3 T cells and subsequently the
algorithm for cell quantification was automatically run. The output data for CD3 quantification were then transferred to GraphPad Prism for analysis of significance. BIOANALYTICS OF SERUM
SAMPLES For PK assessments a specific ELISA was developed for specific analysis of CD3 binding competent drug (active TCB) in the presence of Prot-FOLR1-TCBs. Unbound proteins were washed
away after each step of reagent or sample addition (stepwise assay protocol). An anti CD3-binding site-specific monoclonal antibody was coated onto a microtiter plate followed by sample
addition. Bound analyte was then incubated with digoxigenin-labeled Fc-specific detection antibody (binding to a mutation on the Fc part of the analyte), followed by an incubation with
anti-digoxigenin Fab fragment conjugated to horseradish peroxidase. Finally, formed immobilized immune complexes were visualized by addition of HPPA (3-(4-Hydroxyphenyl)propionic acid)
solution, a fluorogenic POD substrate. The fluorescence intensity was measured with an excitation wavelength of 320 nm (25 nm bandwidth) and an emission wavelength 400 nm (20 nm bandwidth).
Pharmacokinetic evaluation was conducted by noncompartmental methods. Areas under the serum concentration-time curve (AUC) were calculated by linear trapezoidal rule. Bioavailabilities _F_
of the active TCB after Prot-FOLR1-TCB administration were calculated by comparing AUC 0−168 h values of the active FOLR1-TCB following i.v. administration of the respective Prot-FOLR1-TCB
(AUC from pro-TCB) and administration of the active TCB (AUC FOLR1-TCB) according to _F_(%) = (AUC from pro-TCB/AUC TCB) × 100. Dose corrections were not required, as equimolar doses of
Prot-FOLR1-TCB and active FOLR1-TCB were used in the respective studies. STATISTICAL ANALYSIS For statistical analysis of the in vivo TGI curves, one-way ANOVA Tukey−Kramer correction was
used. CD3 T-cell counts were analyzed using two-tailed, unpaired _t_ test using GraphPad Prism 6 Software. _p_ values below 0.05 were considered as significant and were indicated with
asterisks (n.s. _p_ > 0.05, *_p_ ≤ 0.05; **_p_ ≤ 0.01; ***_p_ ≤ 0.001, ****_p_ ≤ 0.0001). Exact _p_ values are shown in the figures. REPORTING SUMMARY Further information on research
design is available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY All data generated or analyzed during this study are included in this published article
(and its supplementary information files). Data underlying Figs. 4, 5, 6, 7, 8a, and 9, Supplementary Figs. 1, 3, and 6 are provided as Source Data file. Material might be obtained for
research purposes only under Material transfer agreement (MTA). REFERENCES * Kobold, S. et al. Immunotherapy in tumors. _Dtsch. Arztebl. Int._ 112, 809–815 (2015). PubMed PubMed Central
Google Scholar * Bacac, M. et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. _Clin. Cancer Res._ 22, 3286–3297 (2016). Article
CAS PubMed Google Scholar * Klein, C. et al. Abstract 3629: Engineering a novel asymmetric head-to-tail 2+1 T-cell bispecific (2+1 TCB) IgG antibody platform with superior T-cell
killing compared to 1+1 asymmetric TCBs. _Cancer Res._ 77, 3629 (2017). Google Scholar * Le Jeune, C. & Thomas, X. Potential for bispecific T-cell engagers: role of blinatumomab in
acute lymphoblastic leukemia. _Drug Des. Dev. Ther._ 10, 757–765 (2016). Google Scholar * Schlothauer, T. et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished
immune effector functions. _Protein Eng. Des. Sel._ 29, 457–466 (2016). Article CAS PubMed Google Scholar * Liddy, N. et al. Monoclonal TCR-redirected tumor cell killing. _Nat. Med._
18, 980–987 (2012). Article CAS PubMed Google Scholar * Hinrichs, C. S. & Restifo, N. P. Reassessing target antigens for adoptive T-cell therapy. _Nat. Biotechnol._ 31, 999–1008
(2013). Article CAS PubMed PubMed Central Google Scholar * Duffy, M. J. Proteases as prognostic markers in cancer. _Clin. Cancer Res._ 2, 613–618 (1996). ADS CAS PubMed Google
Scholar * Tanimoto, H. et al. Ovarian tumor cells express a transmembrane serine protease: a potential candidate for early diagnosis and therapeutic intervention. _Tumour Biol._ 22, 104–114
(2001). Article CAS PubMed Google Scholar * Tanimoto, H. et al. Transmembrane serine protease TADG-15 (ST14/Matriptase/MT-SP1): expression and prognostic value in ovarian cancer. _Br.
J. Cancer_ 92, 278–283 (2005). Article CAS PubMed Google Scholar * Uhland, K. Matriptase and its putative role in cancer. _Cell Mol. Life Sci._ 63, 2968–2978 (2006). Article CAS PubMed
Google Scholar * Oberst, M. et al. Matriptase and HAI-1 are expressed by normal and malignant epithelial cells in vitro and in vivo. _Am. J. Pathol._ 158, 1301–1311 (2001). Article CAS
PubMed PubMed Central Google Scholar * Yu, W., Liu, J., Xiong, X., Ai, Y. & Wang, H. Expression of MMP9 and CD147 in invasive squamous cell carcinoma of the uterine cervix and their
implication. _Pathol. Res. Pract._ 205, 709–715 (2009). Article CAS PubMed Google Scholar * Thant, A. A. et al. Fibronectin activates matrix metalloproteinase-9 secretion via the
MEK1-MAPK and the PI3K-Akt pathways in ovarian cancer cells. _Clin. Exp. Metastasis_ 18, 423–428 (2000). Article CAS PubMed Google Scholar * Schmalfeldt, B. et al. Increased expression
of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. _Clin. Cancer Res._ 7,
2396–2404 (2001). CAS PubMed Google Scholar * Hu, X. et al. Matrix metalloproteinase-9 expression correlates with prognosis and involved in ovarian cancer cell invasion. _Arch. Gynecol.
Obstet._ 286, 1537–1543 (2012). Article CAS PubMed Google Scholar * Yousef, E. M., Tahir, M. R., St-Pierre, Y. & Gaboury, L. A. MMP-9 expression varies according to molecular
subtypes of breast cancer. _BMC Cancer_ 14, 609 (2014). Article PubMed PubMed Central CAS Google Scholar * Sillanpaa, S. et al. Prognostic significance of matrix metalloproteinase-9
(MMP-9) in epithelial ovarian cancer. _Gynecol. Oncol._ 104, 296–303 (2007). Article CAS PubMed Google Scholar * McGowan, P. M. & Duffy, M. J. Matrix metalloproteinase expression and
outcome in patients with breast cancer: analysis of a published database. _Ann. Oncol._ 19, 1566–1572 (2008). Article CAS PubMed Google Scholar * Demeter, A. et al. Molecular prognostic
markers in recurrent and in non-recurrent epithelial ovarian cancer. _Anticancer Res._ 25, 2885–2889 (2005). CAS PubMed Google Scholar * LeBeau, A. M. et al. Imaging a functional
tumorigenic biomarker in the transformed epithelium. _Proc. Natl. Acad. Sci. USA_ 110, 93–98 (2013). Article ADS CAS PubMed Google Scholar * Scaranti, M., Cojocaru, E., Banerjee, S.
& Banerji, U. Exploiting the folate receptor α in oncology. _Nat. Rev. Clin. Oncol._ https://doi.org/10.1038/s41571-020-0339-5 (2020). * Parker, N. et al. Folate receptor expression in
carcinomas and normal tissues determined by a quantitative radioligand binding assay. _Anal. Biochem._ 338, 284–293 (2005). Article CAS PubMed Google Scholar * Giusti, A. M. et al.
Adverse or not adverse—assessment and consequences. In _14th European Congress of Toxicologic Pathology (ESTP)_ (Barcelona, Spain, 2016). * Reiter, Y. et al. Stabilization of the Fv
fragments in recombinant immunotoxins by disulfide bonds engineered into conserved framework regions. _Biochemistry_ 33, 5451–5459 (1994). Article CAS PubMed Google Scholar * Reiter, Y.
et al. Engineering interchain disulfide bonds into conserved framework regions of Fv fragments: improved biochemical characteristics of recombinant immunotoxins containing
disulfide-stabilized Fv. _Protein Eng._ 7, 697–704 (1994). Article CAS PubMed Google Scholar * Glockshuber, R., Malia, M., Pfitzinger, I. & Pluckthun, A. A comparison of strategies
to stabilize immunoglobulin Fv-fragments. _Biochemistry_ 29, 1362–1367 (1990). Article CAS PubMed Google Scholar * Ridgway, J. B. B., Presta, L. G. & Carter, P. ‘Knobs-into-holes’
engineering of antibody CH3 domains for heavy chain heterodimerization. _Protein Eng._ 9, 617–621 (1996). Article CAS PubMed Google Scholar * Klein, C. et al. Progress in overcoming the
chain association issue in bispecific heterodimeric IgG antibodies. _MAbs_ 4, 653–663 (2012). Article PubMed PubMed Central Google Scholar * Schaefer, W. et al. Immunoglobulin domain
crossover as a generic approach for the production of bispecific IgG antibodies. _Proc. Natl. Acad. Sci. USA_ 108, 11187–11192 (2011). Article ADS CAS PubMed PubMed Central Google
Scholar * Ast, O. et al. Bispecific t cell activating antigen binding molecules. WO2016020309 A1, CA2951599A1, US20160075785 (2016). * Kessenbrock, K., Plaks, V. & Werb, Z. Matrix
metalloproteinases: regulators of the tumor microenvironment. _Cell_ 141, 52–67 (2010). Article CAS PubMed PubMed Central Google Scholar * Desnoyers, L. R. et al. Tumor-specific
activation of an EGFR-targeting probody enhances therapeutic index. _Sci. Transl. Med._ 5, 207ra144 (2013). Article PubMed CAS Google Scholar * Norman, D. J. Mechanisms of action and
overview of OKT3. _Ther. Drug Monit._ 17, 615–620 (1995). Article CAS PubMed Google Scholar * Norman, D. J. & Leone, M. R. The role of OKT3 in clinical transplantation. _Pediatr.
Nephrol._ 5, 130–136 (1991). Article CAS PubMed Google Scholar * Kobel, M. et al. Evidence for a time-dependent association between FOLR1 expression and survival from ovarian carcinoma:
implications for clinical testing. An Ovarian Tumour Tissue Analysis consortium study. _Br. J. Cancer_ 111, 2297–2307 (2014). Article CAS PubMed PubMed Central Google Scholar * Konner,
J. A. et al. Farletuzumab, a humanized monoclonal antibody against folate receptor alpha, in epithelial ovarian cancer: a phase I study. _Clin. Cancer Res._ 16, 5288–5295 (2010). Article
CAS PubMed Google Scholar * Ab, O. et al. IMGN853, a Folate Receptor-alpha (FRalpha)-targeting antibody-drug conjugate, exhibits potent targeted antitumor activity against
FRalpha-expressing tumors. _Mol. Cancer Ther._ 14, 1605–1613 (2015). Article CAS PubMed Google Scholar * Armstrong, D. K., White, A. J., Weil, S. C., Phillips, M. & Coleman, R. L.
Farletuzumab (a monoclonal antibody against folate receptor alpha) in relapsed platinum-sensitive ovarian cancer. _Gynecol. Oncol._ 129, 452–458 (2013). Article CAS PubMed Google Scholar
* Tibben, J. G. et al. Pharmacokinetics, biodistribution and biological effects of intravenously administered bispecific monoclonal antibody OC/TR F(ab’)2 in ovarian carcinoma patients.
_Int. J. Cancer_ 66, 477–483 (1996). Article CAS PubMed Google Scholar * Farrell, C. et al. Population pharmacokinetics of farletuzumab, a humanized monoclonal antibody against folate
receptor alpha, in epithelial ovarian cancer. _Cancer Chemother. Pharm._ 70, 727–734 (2012). Article CAS Google Scholar * Autio, K. A., Boni, V., Humphrey, R. W. & Naing, A. Probody
therapeutics: an emerging class of therapies designed to enhance on-target effects with reduced off-tumor toxicity for use in immuno-oncology. _Clin. Cancer Res._
https://doi.org/10.1158/1078-0432.ccr-19-1457 (2019). * Kavanaugh, W. Antibody prodrugs for cancer. _Exp. Opin. Biol. Ther_. https://doi.org/10.1080/14712598.2020.1699053 (2019). * Poreba,
M. Protease-activated prodrugs: strategies, challenges, and future directions. _FEBS J._ https://doi.org/10.1111/febs.15227 (2020). * LaPorte, S. L. et al. Abstract A203: CD3-EGFR bispecific
Probody™ therapeutics induced tumor regressions and increased therapeutic window in preclinical studies. _Mol. Cancer Therapeut._ 14, A203–A203 (2015). Google Scholar * Singh, S. et al.
Abstract 2975: Development of a probody drug conjugate (PDC) targeting CD71 for the treatment of solid tumors and lymphomas. _Cancer Res._ 76, 2975–2975 (2016). Google Scholar * Spira, A.
I. et al. Rizvi9PROCLAIM-CX-072: A First-in-Human Trial to Assess Tolerability of the Protease-Activatable Anti–PD-L1 Probody™ CX-072 in Solid Tumors and Lymphomas Presented at the ASCO 2017
Annual Meeting; June 2–6, 2017; Chicago, Illinois (CytomX Therapeutics, Inc., 2017). * Pai, C. S. et al. Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related
toxicity. _J. Clin. Invest._ 129, 349–363 (2019). Article PubMed Google Scholar * Trang, V. H. et al. A coiled-coil masking domain for selective activation of therapeutic antibodies.
_Nat. Biotechnol._ 37, 761–765 (2019). Article CAS PubMed Google Scholar * Iwahashi, M. et al. CDR substitutions of a humanized monoclonal antibody (CC49): contributions of individual
CDRs to antigen binding and immunogenicity. _Mol. Immunol._ 36, 1079–1091 (1999). Article CAS PubMed Google Scholar * Harding, F. A., Stickler, M. M., Razo, J. & DuBridge, R. B. The
immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. _MAbs_ 2, 256–265 (2010). Article PubMed PubMed Central Google Scholar *
Schmittnaegel, M. et al. Committing cytomegalovirus-specific CD8 T cells to eliminate tumor cells by bifunctional major histocompatibility class I antibody fusion molecules. _Cancer Immunol.
Res._ 3, 764–776 (2015). Article CAS PubMed Google Scholar * List, K., Bugge, T. H. & Szabo, R. Matriptase: potent proteolysis on the cell surface. _Mol. Med._ 12, 1–7 (2006).
Article CAS PubMed PubMed Central Google Scholar * Nomura, T. & Katunuma, N. Involvement of cathepsins in the invasion, metastasis and proliferation of cancer cells. _J. Med.
Invest._ 52, 1–9 (2005). Article PubMed Google Scholar * Mohamed, M. M. & Sloane, B. F. Cysteine cathepsins: multifunctional enzymes in cancer. _Nat. Rev. Cancer_ 6, 764–775 (2006).
Article CAS PubMed Google Scholar * Oberst, M. D. et al. Expression of the serine protease matriptase and its inhibitor HAI-1 in epithelial ovarian cancer: correlation with clinical
outcome and tumor clinicopathological parameters. _Clin. Cancer Res._ 8, 1101–1107 (2002). CAS PubMed Google Scholar * Watermann, I. et al. Activation of CD95L fusion protein prodrugs by
tumor-associated proteases. _Cell Death Differ._ 14, 765–774 (2007). Article CAS PubMed Google Scholar * Condeelis, J. & Pollard, J. W. Macrophages: obligate partners for tumor cell
migration, invasion, and metastasis. _Cell_ 124, 263–266 (2006). Article CAS PubMed Google Scholar * Menen, R. S. et al. Tumor-educated macrophages promote tumor growth and peritoneal
metastasis in an orthotopic nude mouse model of human pancreatic cancer. _Vivo_ 26, 565–569 (2012). Google Scholar * Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next
generation. _Cell_ 144, 646–674 (2011). Article CAS PubMed Google Scholar * Pollard, J. W. Tumour-educated macrophages promote tumour progression and metastasis. _Nat. Rev. Cancer_ 4,
71–78 (2004). Article CAS PubMed Google Scholar * Murphy, G. The ADAMs: signalling scissors in the tumour microenvironment. _Nat. Rev. Cancer_ 8, 929–941 (2008). Article CAS PubMed
Google Scholar * Hessell, A. J. et al. Fc receptor but not complement binding is important in antibody protection against HIV. _Nature_ 449, 101–104 (2007). Article ADS CAS PubMed
Google Scholar * Reid, Y. et al. (2004). * Simms, P. E. & Ellis, T. M. Utility of flow cytometric detection of CD69 expression as a rapid method for determining poly- and oligoclonal
lymphocyte activation. _Clin. Diagn. Lab Immunol._ 3, 301–304 (1996). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank Erwin van
Puijenbroeck, Fabian Birzele, Alexander Bujotzek, Thomas O’Brien, Christa Bayer, Brian Steiner, Inja Waldhauer, Linda Fahrni, Christian Müller, Manuel Späni, Michaela Ketterer, Sara
Colombetti and Marina Bacac for their help. S.K. and S.E. are supported by grants from the Wilhelm Sander Stiftung (grant number 2014.018.1 to S.E. and S.K.), the international doctoral
program “i-Target: Immunotargeting of cancer” funded by the Elite Network of Bavaria (to S.K. and S.E.), the Melanoma Research Alliance (grant number N269626 to S.E. and 409510 to S.K.), the
Marie-Sklodowska-Curie “Training Network for the Immunotherapy of Cancer (IMMUTRAIN)” funded by the H2020 program of the European Union (to S.E. and S.K.), the Else
Kröner-Fresenius-Stiftung (to S.K.), the German Cancer Aid (to S.K.), the Ernst-Jung-Stiftung (to S.K.), by LMU Munich’s Institutional Strategy LMUexcellent within the framework of the
German Excellence Initiative (to S.E. and S.K.), the Bundesministerium für Bildung und Forschung (project Oncoattract to S.E. and S.K.), the Deutsche Forschungsgemeinschaft, the
José-Carreras Leukämie Stiftung, the Hector-Foundation (all to S.K.) and the European Research Council (ERC, grant 756017, ARMOR-T to S.K.). AUTHOR INFORMATION Author notes * Jigar Patel
& Eric Sullivan Present address: Nimble Therapeutics Inc., 500S Rosa Rd, Madison, WI, 53719, USA * These authors contributed equally: Peter Brünker, Christian Klein. AUTHORS AND
AFFILIATIONS * Roche Pharma Research & Early Development, Roche Innovation Center Zurich, Wagistrasse 10, 8952, Schlieren, Switzerland Martina Geiger, Johannes Sam, Valeria Nicolini,
Anne Freimoser-Grundschober, Sandra Grau-Richards, Pablo Umaña, Peter Brünker & Christian Klein * Center of Integrated Protein Science Munich (CIPS-M) and Division of Clinical
Pharmacology, Department of Medicine IV, Klinikum der Universität München, Lindwurmstraße 2a, Member of the German Center for Lung Research (DZL), 80337, Munich, Germany Martina Geiger,
Stefan Endres & Sebastian Kobold * Roche Pharma Research & Early Development, Roche Innovation Center Munich, Nonnenwald 2, 82372, Penzberg, Germany Kay-Gunnar Stubenrauch, Gregor
Jordan, Jan Eckmann & Carina Hage * Roche Pharma Research & Early Development, Roche Innovation Center Basel, Grenzacherstrasse 124, 4070, Basel, Switzerland Wolfgang F. Richter
& Matthias E. Lauer * Roche Diagnostics, CPS Research and Development, Nonnenwald 2, 82372, Penzberg, Germany Mirko Ritter * Center for Cellular Imaging and Nano Analytics, Biozentrum,
University of Basel, 4070, Basel, Switzerland Henning Stahlberg & Philippe Ringler * Roche Sequencing, NimbleGen, Madison, WI, 53719, USA Jigar Patel & Eric Sullivan * Einheit für
Klinische Pharmakologie (EKLiP), Helmholtz Zentrum München, German Research Center for Environmental Health (HMGU), Neuherberg, Germany Stefan Endres & Sebastian Kobold * German Center
for Translational Cancer Research (DKTK), Partner Site Munich, Munich, Germany Stefan Endres & Sebastian Kobold Authors * Martina Geiger View author publications You can also search for
this author inPubMed Google Scholar * Kay-Gunnar Stubenrauch View author publications You can also search for this author inPubMed Google Scholar * Johannes Sam View author publications You
can also search for this author inPubMed Google Scholar * Wolfgang F. Richter View author publications You can also search for this author inPubMed Google Scholar * Gregor Jordan View author
publications You can also search for this author inPubMed Google Scholar * Jan Eckmann View author publications You can also search for this author inPubMed Google Scholar * Carina Hage
View author publications You can also search for this author inPubMed Google Scholar * Valeria Nicolini View author publications You can also search for this author inPubMed Google Scholar *
Anne Freimoser-Grundschober View author publications You can also search for this author inPubMed Google Scholar * Mirko Ritter View author publications You can also search for this author
inPubMed Google Scholar * Matthias E. Lauer View author publications You can also search for this author inPubMed Google Scholar * Henning Stahlberg View author publications You can also
search for this author inPubMed Google Scholar * Philippe Ringler View author publications You can also search for this author inPubMed Google Scholar * Jigar Patel View author publications
You can also search for this author inPubMed Google Scholar * Eric Sullivan View author publications You can also search for this author inPubMed Google Scholar * Sandra Grau-Richards View
author publications You can also search for this author inPubMed Google Scholar * Stefan Endres View author publications You can also search for this author inPubMed Google Scholar *
Sebastian Kobold View author publications You can also search for this author inPubMed Google Scholar * Pablo Umaña View author publications You can also search for this author inPubMed
Google Scholar * Peter Brünker View author publications You can also search for this author inPubMed Google Scholar * Christian Klein View author publications You can also search for this
author inPubMed Google Scholar CONTRIBUTIONS Conception and design: M.G., P.U., C.K. and P.B. Development of methodology and data acquisition: M.G., W.F.R., G.J.; S.G.-R., A.F.-G., J.P.,
E.S., M.E.L., K.-G.S., M.R., V.N., H.S. and P.R. Analysis and interpretation of data: M.G., C.K., P.B., W.F.R., J.S., V.N., C.H., J.E., A.F.-G., M.E.L., M.R., H.S. and P.R. Writing and
review of the manuscript: M.G., C.K., P.B., J.S., W.F.R., A.F.-G., S.K. Administrative, technical, or material support: M.G., K.-G.S., M.R. Study supervision: C.K., P.B., S.E., S.K., P.U.
CORRESPONDING AUTHOR Correspondence to Christian Klein. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare the following competing interests. Parts of this work have been performed
for the doctoral thesis of M.G. associated to the international doctoral program “i-Target” at the Ludwig-Maximilians-Universität München. M.G., K.-G.S., A.F.-G., M.R., M.E.L., J.S., J.E.,
C.H., W.F.R., G.J., V.N., P.U., P.B. and C.K. are employees of Roche. J.P. and E.S. are employees and hold ownership in Nimble Therapeutics. J.P. and E.S. own Nimble Therapeutics stock.
C.K., P.B., A.F.-G., P.U., K.-G.S., M.G., E.S., J.P. are inventors in patent applications related to this work. C.K., P.U., M.G., P.B., W.F.R., M.R., G.J., S.G.R., J.S., A.F.-G. own Roche
stock. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports
are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International
License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source,
provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons
license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Geiger, M., Stubenrauch, KG., Sam, J. _et al._ Protease-activation using
anti-idiotypic masks enables tumor specificity of a folate receptor 1-T cell bispecific antibody. _Nat Commun_ 11, 3196 (2020). https://doi.org/10.1038/s41467-020-16838-w Download citation *
Received: 25 January 2018 * Accepted: 29 May 2020 * Published: 24 June 2020 * DOI: https://doi.org/10.1038/s41467-020-16838-w SHARE THIS ARTICLE Anyone you share the following link with
will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative
Trending News
404 Not Found!You're using an Ad-Blocker. BeforeItsNews only exists through ads.We ask all patriots who appreciate the evil we expose ...
The page you were looking for doesn't exist.You may have mistyped the address or the page may have moved.By proceeding, you agree to our Terms & Conditions and our ...
404 Not Found!You're using an Ad-Blocker. BeforeItsNews only exists through ads.We ask all patriots who appreciate the evil we expose ...
Transmission of cml or of t(9; 22) and bcr/abl? They are not the sameAccess through your institution Buy or subscribe In a recent Letter to the Editor de Brito _et al._1 describe a transpla...
Coronavirus anxiety could be causing your weird dreamsDavid Salkeld needs sleep. The 54-year-old registered nurse works up to 60 hours a week in a North Carolina emergency ro...
Latests News
Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-t cell bispecific antibodyABSTRACT T-cell bispecific antibodies (TCBs) crosslink tumor and T-cells to induce tumor cell killing. While TCBs are ve...
New claimants want to reopen rampur’s 47-year-old property dispute again - times of indiaRampur family: Nawab Raza Ali Khan in extreme right (in cap) with his sister Nawabzadi Kulsoom Begum in white in the mid...
The page you were looking for doesn't exist.You may have mistyped the address or the page may have moved.By proceeding, you agree to our Terms & Conditions and our ...
Javascript support required...
Accomplish your financial goals with a fresh startMemorial Day Sale! Join AARP for just $11 per year with a 5-year membership Join now and get a FREE gift. Expires 6/4 G...