A novel splicing mutation in slc9a6 in a boy with christianson syndrome
A novel splicing mutation in slc9a6 in a boy with christianson syndrome"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT A loss of function mutation in _SLC9A6_ (Xq26.3) is responsible for Christianson syndrome in males. We identified a novel splicing mutation (NM_006359.2:c.1141-8C>A) of _SLC9A6_
in a seven-year-old boy with microcephaly, severe developmental delay, and intractable epilepsy. Functional analysis found multiple aberrant transcripts, none of which maintained the
canonical open reading frame. Computer prediction tools, however, failed to detect all of the aberrant transcripts. A loss of function mutation in the _SLC9A6_ gene (Xq26.3) is responsible
for Christianson syndrome (CS), which is characterized by severe global developmental delay, developmental regression, acquired microcephaly, intractable epilepsy, ataxia, ophthalmoplegia,
and sometimes, death at a young age1,2. The clinical features of CS overlap with those of Angelman syndrome (AS), which is caused by a lack of expression of the maternally inherited _UBE3A_
gene located on 15q11.23. _SLC9A6_ encodes the Na+/H+ exchanger protein NHE6. This protein regulates the luminal pH of early and recycling endosomes involved in the trafficking of proteins
essential for structural and functional plasticity at glutamatergic synapses4. NHE6 has an important role in the growth of dendritic spines and the development of normal brain wiring5. Here,
we identified a novel _SLC9A6_ splicing mutation in a seven-year-old boy with microcephaly, severe developmental delay, and intractable epilepsy. To evaluate the mutation, we used various
computer prediction tools as well as reverse transcription polymerase chain reaction (RT-PCR) and cloning to assess transcripts and confirm the pathogenicity of the mutation. The case study,
a seven-year-old Japanese boy, was born at term with a birth weight of 2978 g (−0.4 SD), length of 50.2 cm (+0.4 SD), and head circumference of 31.4 cm (−1.5 SD). His development delayed
gradually, achieving head control at four months, sitting at nine months, and pulling to stand at two years. Currently, he cannot stand independently nor speak meaningful words. At 10 months
of age, he developed intractable seizures of variable types: tonic-clonic convulsion, impairment of consciousness, focal seizure, and epileptic negative myoclonus. He was treated with
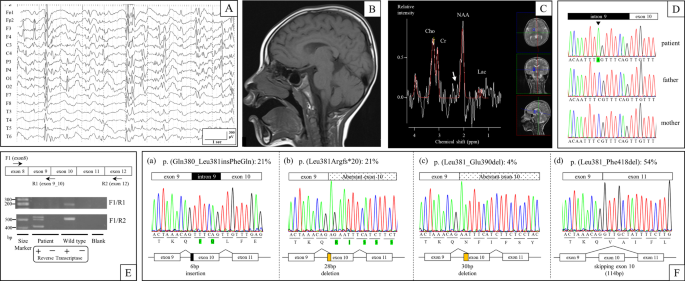
multiple antiepileptic drugs that had insufficient therapeutic effects. An electroencephalography (EEG) performed at four years showed focal epileptic discharges with generalization in
multiple foci (Fig. 1a). At four years of age, his weight was 14.5 kg (−0.7 SD), length was 102 cm (−0.1 SD), and head circumference was 46.2 cm (−2.6 SD), indicating microcephaly. Brain
magnetic resonance imaging (MRI) (performed at 1, 2, and 4 years of age) and magnetic resonance spectroscopy (MRS) (performed at 4 years of age) showed no abnormal findings (Fig. 1b, c). At
five years of age, we suspected the diagnosis of AS due to severe developmental delay, trunk ataxia, intractable seizures, microcephaly, and frequent smiling. We first performed genetic
tests, including fluorescent in situ hybridization (FISH), DNA methylation, and mutation analysis of the protein coding exons of _UBE3A_ by Sanger sequencing, but all of the tests were
normal. Next, we performed targeted next-generation sequencing with the Ion Torrent Personal Genome Machine system (Life Technologies, Carlsbad, California). An amplicon library of the
target exons and flanking sequence was prepared with the use of an Ion AmpliSeq Custom Panel (Life Technologies), which included _UBE3A_, _SLC9A6_, _TCF4_, _MBD5_, _CDKL5_, _MECP2_, and
_FOXG1_. Sequence analysis pipelines were established with use of the workflow in CLC Genomic Workbench 7.0 (CLC bio, Aarhus, Denmark). We identified a _de novo_ hemizygous splicing mutation
(c.1141-8C>A) in _SLC9A6_ (NM_006359.2), which was confirmed by Sanger sequencing using the _SLC9A6_-intron 9-Fwd (5’-TCCACATTTGCTCCCTTCT-3’) and _SLC9A6_-exon 10-Rev
(5’-ACCACATACTCAAAACCCAC-3’) primer pair (Fig. 1d). We predicted that the mutation affected RNA splicing because it resulted in a new AG acceptor site six nucleotides upstream of the
canonical acceptor site of exon 10. To evaluate the mutation, we used multiple computer prediction tools. CRYP-SKIP (http://cryp-skip.img.cas.cz/) provides an overall probability of cryptic
splice-site activation (as opposed to exon skipping) termed _P_CR-E6. _P_CR-E calculates a value between 0 and 1, and lower values favor exon skipping. The _P_CR-E prediction score for
_SLC9A6_ (c.1141-8C>A) was 0.20, thus favoring exon skipping. Next, we used Alamut Visual software (version 2.10, Interactive Biosoftware, Rouen, France), which assesses genomic sequences
(wild type and mutant) using five splicing prediction tools (SpliceSiteFinder-like, MaxEntScan, Neural Network Splice, GeneSplicer, and Human Splicing Finder) based on different
algorithms7. All five algorithms predicted a strength reduction in the canonical acceptor site. The prediction scores for the aberrant acceptor site (six nucleotides upstream of the
canonical acceptor site) increased with three algorithms, and the scores did not change at any point downstream of the canonical acceptor site (Table 1). To confirm the RNA splicing results,
we performed RT-PCR, cloning, and Sanger sequencing using total RNA from Epstein-Barr virus-induced lymphoblastoid cell lines established from peripheral leukocytes. RT-PCR using the
_SLC9A6_-exon 8-Fwd1 (5′-ACCAAATTACGGGAGTTCCA-3′) and _SLC9A6_-exon 12-Rev2 (5′-CACCACCAAATACCCACAC-3′) primer pair revealed the presence of multiple transcripts (Fig. 1e). _SLC9A6_ cDNA was
then ligated into a TOPO cloning vector (Life Technologies) and transformed into TOP10 Competent Cells (Life Technologies), and 24 colonies were screened by extracting plasmid DNA using a
QIAprep Spin Miniprep Kit (Qiagen, Hilden, Germany). Sanger sequencing of the plasmid clones identified four unique, aberrant transcripts but no canonical transcripts. Of the 24 cDNA
transcripts screened, five (21%) had the six-nucleotide addition of the intronic sequence to the 5’ end of exon 10 that was predicted by our in silico analysis (Fig. 1f, transcript (a)). We
also identified five (21%) transcripts and one (4%) transcript with 5’ exon 10 deletions of 28 and 30 nucleotides, respectively, as well as 13 transcripts (54%) with complete skipping of
exon 10 (Fig. 1f, transcripts (b), (c), and (d)). Furthermore, we performed RT-PCR using the _SLC9A6_-exon 8-Fwd1 and _SLC9A6_-exon9_10-Rev1 (5′-GCTCAAACAACTGTTTAGTTCTA-3′) primer pair,
which amplified only canonical transcripts, and it revealed canonical transcripts in control DNA but no amplification in that of the patient. CS was first reported in 1999 in a Caucasian
South African family with multiple affected males presenting with severe intellectual disability, mutism despite apparently normal hearing, intractable epilepsy, and limited life
expectancy1. As some of the clinical features of CS are shared with AS, 1.8–5.5% patients with AS-like phenotypes have _SLC9A6_ mutations3,8. The characteristic features that distinguish CS
from AS are external ophthalmoplegia, developmental regression with loss of motor skills, progressive atrophy of the inferior cerebellar vermis, and an increased glutamine-glutamate peak in
the basal ganglia on MRS9. Our patient, however, did not show any of these characteristic features at seven years of age. Pescosolido et al. reported that CS patients had regression in
walking (57%), eating (14%), loss of few words/sounds (57%), eye contact/facial expressions (14%) and other fine/gross motor skills (14%) after a medical illness and/or seizure cluster10;
therefore, we intend to follow our patient carefully. Using target sequencing, we identified a _de novo_ hemizygous intronic mutation (c.1141-8C>A) in _SLC9A6_ (NM_006359.2), which
resulted in a new AG acceptor site six nucleotides upstream of the canonical acceptor site of exon 10. In silico computer prediction analysis was performed prior to functional analysis of
the mutation. CRYP-SKIP predicted the mutation would tend to cause exon skipping. Alamut visual predicted a decreased score for the canonical acceptor site of exon 10 in all five algorithms
and an increased score for c.1141-6, which is adjacent to the aberrant AG acceptor site, in 3 of the 5 algorithms. To confirm the differential RNA splicing caused by the intronic mutation,
we performed functional analyses using RT-PCR, cloning, and Sanger sequencing. We found multiple aberrant transcripts in _SLC9A6_ involving exon 10, but no canonical transcripts were
identified. Twenty-one percent of transcripts had the six-nucleotide addition of the intronic sequence to the 5′-end of exon 10, as predicted by our in silico analysis, which leads to a
two-amino-acid insertion (p.(Gln380_Leu381insPheGln)) that we termed transcript (a). Transcripts (b) and (c) had 5′ exon 10 deletions of 28 and 30 nucleotides occurring in 21 and 4% of
transcripts, respectively, leading to a p.(Leu381Argfs*20) frameshift in transcript (b) and a 10-amino-acid deletion (p.(Leu381_Glu390del)) in transcript (c). Fifty-four percent of
transcripts had complete skipping of exon 10, termed transcript (d), due to a 114-bp deletion that led to a 38-amino-acid deletion (p.(Leu381_Phe418del)). Exon 10 in _SLC9A6_ encodes part of
the functional domain that interacts with angiotensin II type 2 receptor11. Transcripts (b) and (d) (frameshift mutation and single exon deletion, respectively) are likely to disrupt gene
function. The functional consequences of transcripts (a) and (c) (small in-frame insertion/deletion) are unclear as they are also located in the functional domain and only correspond to 25%
of transcripts. Since the phenotype of the patient is consistent with CS, we conclude that not enough functional transcripts of _SLC9A6_ are being expressed, and the c.1141-8C>A mutation
is pathogenic. Comparing computer predictions to RNA transcript analysis, transcripts (a) and (d) were predicted by Alamut visual and CRYP-SKIP, respectively, but transcripts (b) and (c)
were not predicted. Previous studies comparing the functional consequences of splice site mutations in _HR_ (using CRYP-SKIP)12 and _MYBPC3_, _ACTC1_, and _SCN5A_ (using Alamut analysis)7
concluded that prediction programs underestimate the impact of intronic mutations and that functional analyses, such as RT-PCR and minigene analysis, are necessary. In our experience,
computer prediction tools predicted two of the four aberrant transcripts detected by RT-PCR, highlighting the need to develop more accurate computer prediction tools. HGV DATABASE The
relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2543 REFERENCES * Christianson, A. L. et al. X linked severe
mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large South African kindred is localised to Xq24-q27. _J. Med. Genet._ 36, 759–766
(1999). Article CAS Google Scholar * Schroer, R. J. et al. Natural history of Christianson syndrome. _Am. J. Med. Genet. A._ 152A, 2775–2783 (2010). Article Google Scholar * Gilfillan,
G. D. et al. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. _Am. J. Hum. Genet._ 82, 1003–1010 (2008).
Article CAS Google Scholar * Deane, E. C. et al. Enhanced recruitment of endosomal Na+/H+ exchanger NHE6 into Dendritic spines of hippocampal pyramidal neurons during NMDA
receptor-dependent long-term potentiation. _J. Neurosci._ 33, 595–610 (2013). Article CAS Google Scholar * Takahashi, Y. et al. A loss-of-function mutation in the SLC9A6 gene causes
X-linked mental retardation resembling Angelman syndrome. _Am. J. Med. Genet. B. Neuropsychiatr. Genet._ 156B, 799–807 (2011). Article Google Scholar * Divina, P., Kvitkovicova, A.,
Buratti, E. & Vorechovsky, I. Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. _Eur. J. Hum. Genet._ 17, 759–765 (2009). Article CAS Google
Scholar * Frisso, G. et al. Functional studies and in silico analyses to evaluate non-coding variants in inherited cardiomyopathies. _Int. J. Mol. Sci._ 17, 1883 (2016). Article Google
Scholar * Fichou, Y. et al. Mutation in the SLC9A6 gene is not a frequent cause of sporadic Angelman-like syndrome. _Eur. J. Hum. Genet._ 17, 1378–1380 (2009). Article Google Scholar *
Tan, W.-H., Bird, L. M., Thibert, R. L. & Williams, C. A. If not Angelman, what is it? a review of Angelman-like syndromes. _Am. J. Med. Genet. A._ 164A, 975–992 (2014). Article Google
Scholar * Pescosolido, M. F. et al. Genetic and phenotypic diversity of NHE6 mutations in Christianson syndrome. _Ann. Neurol._ 76, 581–593 (2014). Article CAS Google Scholar * Zanni, G.
et al. A novel mutation in the endosomal Na+/H+ exchanger NHE6 (SLC9A6) causes Christianson syndrome with electrical status epilepticus during slow-wave sleep (ESES). _Epilepsy Res._ 108,
811–815 (2014). Article CAS Google Scholar * Refke, M. et al. Functional analysis of splice site mutations in the human hairless (HR) gene using a minigene assay. _Br. J. Dermatol._ 165,
1127–1132 (2011). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank the patient and family who participated in our study. AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Department of Pediatrics and Neonatology, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan Daisuke Ieda, Ikumi Hori, Yuji Nakamura, Kei Ohashi, Yutaka
Negishi, Ayako Hattori & Shinji Saitoh * Department of Pediatrics, Tokyo-Kita Medical Center, Tokyo, Japan Atsuko Arisaka & Setsuko Hasegawa * Department of Pediatrics and
Developmental Biology, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo, Japan Setsuko Hasegawa Authors * Daisuke Ieda View author publications You
can also search for this author inPubMed Google Scholar * Ikumi Hori View author publications You can also search for this author inPubMed Google Scholar * Yuji Nakamura View author
publications You can also search for this author inPubMed Google Scholar * Kei Ohashi View author publications You can also search for this author inPubMed Google Scholar * Yutaka Negishi
View author publications You can also search for this author inPubMed Google Scholar * Ayako Hattori View author publications You can also search for this author inPubMed Google Scholar *
Atsuko Arisaka View author publications You can also search for this author inPubMed Google Scholar * Setsuko Hasegawa View author publications You can also search for this author inPubMed
Google Scholar * Shinji Saitoh View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Shinji Saitoh. ETHICS DECLARATIONS
CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer Nature remains neutral with regard to jurisdictional claims
in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ieda, D., Hori, I., Nakamura, Y. _et al._ A novel splicing mutation in _SLC9A6_ in a boy with Christianson syndrome. _Hum Genome Var_ 6, 15
(2019). https://doi.org/10.1038/s41439-019-0046-x Download citation * Received: 06 August 2018 * Revised: 10 January 2019 * Accepted: 18 February 2019 * Published: 25 March 2019 * DOI:
https://doi.org/10.1038/s41439-019-0046-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Real madrid will call tottenham boss mauricio pochettino - balagueZidane resigned as head coach of Real Madrid just days after leading the Spanish club to a third straight Champions Leag...
Were team gb’s skeleton suits responsible for fantastic three medal haul?Team GB skeleton rider Lizzie Yarnold won a stunning Winter Olympic gold on February 17, backed up by bronzes for Laura ...
Kim jong-un’s brother ‘at risk of assassination’KIM JONG-UN ATTENDS AN EMERGENCY POLITBURO MEETING Supreme Leader Kim Jong-un appeared for the first time since passing ...
Comment: weeds can make a garden cheaper and more manageable in franceCOLUMNIST SAMANTHA DAVID TELLS HOW EMBRACING NATURE HAS HIDDEN BENEFITS The day after I moved into my French farmhouse, ...
Certificates programs can boost your career and skillsDerrick Lewis has new appreciation for something a coworker told him 20 years ago: “You have to keep learning.” That adv...
Latests News
A novel splicing mutation in slc9a6 in a boy with christianson syndromeABSTRACT A loss of function mutation in _SLC9A6_ (Xq26.3) is responsible for Christianson syndrome in males. We identifi...
Developmental gene networks: a triathlon on the course to t cell identityKEY POINTS * T cell development depends on the regulated progression of progenitor cells through three major phases that...
A confused bald eagle landed on a fan at the Cotton Bowl and it was impossible not to think of James Paxton By Adrian Garro • December 29, 2018 TaijuaA confused bald eagle landed on a fan at the Cotton Bowl and it was impossible not to think of James Paxton By Adrian Ga...
1662 indomethacin, immature fetal pulmonary vascular resistance (pvr), and lung inflationABSTRACT Indomethacin (I) was studied in the isolated, perfused immature (27 days' gestation) fetal rabbit lung. Lu...
Javascript support required...