Genomic and geographic footprints of differential introgression between two divergent fish species (solea spp. )
Genomic and geographic footprints of differential introgression between two divergent fish species (solea spp. )"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Investigating gene flow between closely related species and its variation across the genome is important to understand how reproductive barriers shape genome divergence before
speciation is complete. An efficient way to characterize differential gene flow is to study how the genetic interactions that take place in hybrid zones selectively filter gene exchange
between species, leading to heterogeneous genome divergence. In the present study, genome-wide divergence and introgression patterns were investigated between two sole species, _Solea
senegalensis_ and _Solea aegyptiaca_, using restriction-associated DNA sequencing (RAD-Seq) to analyze samples taken from a transect spanning the hybrid zone. An integrative approach
combining geographic and genomic clines methods with an analysis of individual locus introgression accounting for the demographic history of divergence was conducted. Our results showed that
the two sole species have come into secondary contact postglacially, after experiencing a prolonged period (_ca_. 1.1 to 1.8 Myrs) of allopatric separation. Secondary contact resulted in
the formation of a tension zone characterized by strong reproductive isolation, which only allowed introgression in a limited fraction of the genome. We found multiple evidence for a
preferential direction of introgression in the _S. aegyptiaca_ genetic background, indicating a possible recent or ongoing movement of the hybrid zone. Deviant introgression signals found in
the opposite direction suggested that _S. senegalensis_ could have possibly undergone adaptive introgression that has not yet spread throughout the entire species range. Our study thus
illustrates the varied outcomes of genetic interactions between divergent gene pools that recently met after a long history of divergence. You have full access to this article via your
institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS VARIABLE LEVELS OF INTROGRESSION BETWEEN THE ENDANGERED _PODARCIS CARBONELLI_ AND HIGHLY DIVERGENT CONGENERIC SPECIES Article
16 November 2020 GENOMIC DATA AND MULTI-SPECIES DEMOGRAPHIC MODELLING UNCOVER PAST HYBRIDIZATION BETWEEN CURRENTLY ALLOPATRIC FRESHWATER SPECIES Article 30 August 2021 POPULATION GENETICS
REVEALS DIVERGENT LINEAGES AND ONGOING HYBRIDIZATION IN A DECLINING MIGRATORY FISH SPECIES COMPLEX Article 04 June 2022 INTRODUCTION Reproductive isolation and hybridization are antinomic
but related processes that are tuned by the same evolutionary forces. Natural hybridization between two divergent lineages occurs as long as reproductive isolation is not totally established
(Dobzhansky 1937; Mayr 1942; Coyne and Orr 2004). The resulting exchange of genetic material beyond the first hybrid generation occurs through the production of backcross and recombinant
genotypes that are exposed to selection, allowing neutral and advantageous alleles to spread among divergent lineages but preventing gene flow at the loci involved in reproductive isolation
(Barton 1979). Hybrid zones, the geographic areas where hybridization occurs, therefore act as selective filters which allow us to unravel the strength of reproductive barriers and their
impact on the patterns and dynamics of gene exchanges at various stage of speciation (Barton and Hewitt 1985; Barton and Bengtsson 1986; Hewitt 1988; Martinsen et al. 2001). The
semipermeable nature of species boundaries was first evidenced from differential introgression patterns in hybrid zones (Harrison 1990), that is, variation among loci in the level of
incorporation of alleles from one lineage into the other (Payseur 2010; Harrison and Larson 2016). Introgression at a given neutral marker depends on the antagonistic effects between
counter-selection on a nearby selected locus and recombination with that barrier locus (Barton and Bengtsson 1986). Therefore, differential gene flow across the genome reflects both
recombinational distance to a barrier locus and selection acting on that locus. Different methods have been developed to detect individual loci with introgression behaviors departing from
the genome-wide average. This includes the analysis of spatial allele frequency patterns in a transect spanning the hybrid zone, or variation in ancestry proportion at individual loci
compared to genome-wide expectations in admixed genotypes (see Payseur 2010 for a review). The geographic cline method is a powerful approach to analyze the relationship between allele
frequency and geographic distance to the center of a hybrid zone (Endler 1977; Barton and Hewitt 1985; Barton and Gale 1993; Porter et al. 1997; Teeter et al. 2008). Fitting a cline model to
observed allele frequency patterns allows inferring fundamental parameters such as cline center, dispersal, and the strength of selection. Therefore, important information concerning the
balance between migration and selection can be obtained using the geographic cline method, such as the identification of which loci tend to introgress neutrally and which do not. The genomic
cline method is a related approach developed to deal with hybrid zones that are not clinally structured, such as mosaic hybrid zones (Harrison and Rand 1989; Bierne et al. 2003; Larson et
al. 2013). The change in allele frequency at individual loci is analyzed along a gradient of genomic admixture instead of a spatial gradient, enabling a comparison of individual locus
introgression patterns with regard to the genome-wide average pattern of admixture (Gompert and Buerkle 2009, 2011; Fitzpatrick 2013). As for geographic clines, this genomic cline method
also provides a quantitative assessment of the excess of ancestry to a given parental species, as well as the rate of change in allele frequency from one species to the other along the
admixture gradient. Although both approaches are useful to document the effect of selection and recombination on differential introgression in admixed genotypes, they do not specifically
account for the demographic history of the parental species (Payseur and Rieseberg 2016). An alternative approach that partly overcomes this limitation is the use of demo-genetic models that
account for varying rates of introgression among loci during the divergence history. Such methods for reconstructing the history of gene flow between semi-isolated lineages have been
developed within Bayesian (Sousa et al. 2013), approximate Bayesian computation (Roux et al. 2013, 2016), or approximate likelihood frameworks (Tine et al. 2014; Le Moan et al. 2016; Rougeux
et al. 2017). Using summary statistics like the joint allelic frequency spectrum, which depicts correlations in allele frequencies between lineages outside the hybrid zone, they capture
variable behaviors among loci and allow quantifying the degree of semi-permeability reflecting the overall balance between gene flow and selection. These methods have the advantage of
specifically accounting for the history of gene flow during divergence, using contrasted speciation scenarios such as primary differentiation, ancient migration (AM), or secondary contact
(SC). However, they do not provide information about individual locus behavior as the cline methods do. Here, we pushed these inference methods one step further in order to assess the
probability for a given locus to belong to one of two categories: (i) loci with a reduced effective migration rate due to selection and linkage, and (ii) loci which can readily introgress.
The flatfish species _Solea senegalensis_ and _Solea aegyptiaca_ are two economically important species in the Mediterranean basin that hybridize along the northern Tunisian coasts, where
they form a hybrid zone (She et al. 1987). Mitochondrial divergence between them is high (~2%) relative to other vertebrate species pairs that also hybridize in nature, such as the house
mice _Mus m. musculus_ and _M. m. domesticus_ (0.3%, based on sequences retrieved from Genebank) or Atlantic and Mediterranean lineages of _Dicentrarchus labrax_ (0.7%, Tine et al. 2014).
Based on an analysis of spatial allele frequency patterns at a dozen of allozymic and intronic loci, Ouanes et al. (2011) proposed that the hybrid zone between _S. senegalensis_ and _S.
aegyptiaca_ was centered in Bizerte lagoon, acting as a non-stable unimodal tension zone stemming from a SC. They also suggested that the zone could have undergone a recent expansion.
Recently, Souissi et al. (2017) showed the existence of morphological transgressions within the contact zone, possibly indicating a reduced fitness of recombined compared to parental
genotypes. However, the low number of markers in these latter studies could not provide a clear description of the genetic exchanges across the genome of these two species, nor quantifying
the strength of the forces maintaining the incomplete reproductive isolation between them. In the present work, high-throughput genotyping using restriction-associated DNA (RAD) sequencing
(Baird et al. 2008) was carried out in individuals sampled at both sides of the hybrid zone, with a particular effort in the contact zone itself. By combining three different methods
(genomic and geographic cline analysis and historical demographic inference) exploiting different aspects of the data, we provide new insights on the history of divergence between _S.
senegalensis_ and _S. aegyptiaca_, and a genome-wide description of varied patterns of introgression attributed to recent or ongoing movement of the hybrid zone. MATERIALS AND METHODS
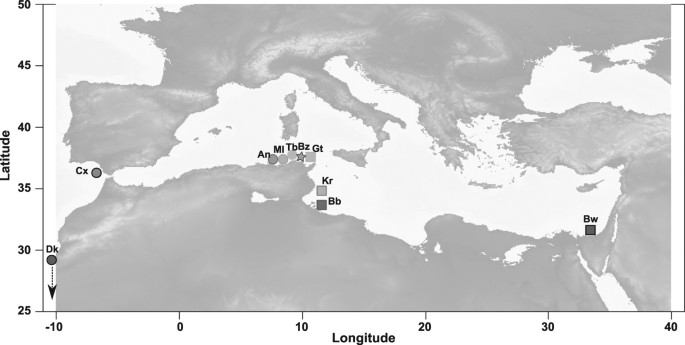
SAMPLING A total of 161 samples were collected from 10 locations spanning the distribution range of _Solea senegalensis_ and _S. aegyptiaca_ from Senegal to Egypt (Fig. 1). This sampling
strategy aimed at covering the geographical distribution of both species, including a detailed transect of their natural hybrid zone in Tunisia. Five sampling locations were collected
throughout the _S. senegalensis_ parental zone, two in the Atlantic Ocean (10 individuals from Dakar in Senegal and 16 from the Gulf of Cadiz in Spain), and three in the Western
Mediterranean Sea (15 individuals from Annaba and 8 from Mellah Lagoon in Algeria, and 10 from Tabarka in Tunisia). Three locations were sampled across the geographical range of _S.
aegyptiaca_ in the Eastern Mediterranean Sea (13 individuals from Kerkennah Island and 15 from El Biban lagoon in Tunisia, and 20 samples from Bardawil lagoon in Egypt). Sampling size was
locally increased in the Tunisian region where both species coexist and hybridization has been reported (Bizerte lagoon _n_ = 29 and Gulf of Tunis _n_ = 25). Finally, a total of seven
individuals belonging to the closely related species _S. solea_ were sampled in Tunisia to provide an outgroup species for the orientation of ancestral and derived alleles in _S.
senegalensis_ and _S. aegyptiaca_. RAD LIBRARY PREPARATION AND SEQUENCING Whole-genomic DNA was extracted from fin clips using the DNeasy Blood & Tissue kit (Qiagen). The presence of
high molecular weight DNA was checked on a 1% agarose gel, and double-stranded DNA concentration was quantified using Qubit 2.0 and standardized to 25 ng per μl. RAD library construction
followed a modified version of the original single-end RAD-Seq protocol (Baird et al. 2008). Briefly, 1 μg of genomic DNA from each individual was digested using the restriction enzyme
_Sbf_I-HF (NEB), and ligated to one of 32 unique molecular barcodes of 5–6 bp. Ligated products were then combined in equimolar proportions into six RAD libraries, each made of a multiplex
of 32 individuals originating from various localities. Each library was finally sequenced in 101 bp single read mode on a separate lane of an Illumina HiSeq2500 sequencer, at the sequencing
platform “Génomique Intégrative et Modélisation des Maladies Métaboliques” (UMR 8199, Lille, France). BIOINFORMATIC ANALYSES Raw reads were de-multiplexed based on individual barcode
information and subsequently end-clipped to 95 bp to homogenize read length after removing barcodes of different lengths. Read quality filtering was performed using the sliding window
approach implemented in the module _process_radtags_ from the _stacks_ pipeline (Catchen et al. 2013). This allowed us to exclude reads in which the average quality of 15 adjacent bases fell
below a raw phred score of 10. Retained read were then aligned to a draft assembly of the _S. senegalensis_ genome (98,590 scaffolds, total length 740 Mb, N50 contig length 10,767 bp,
Manchado et al. 2016) using _bowtie 2_v.2.1.0 (Langmead and Salzberg 2012) with the --very-sensitive option in --end-to-end mode. In order to take into account both the level of divergence
among species and the possibility of introgression while accounting for hidden paralogy, we used a subset of individuals to empirically determine the optimal maximum number of mismatches
allowed between aligned reads and the reference genome, following a procedure described previously (Le Moan et al. 2016; Rougemont et al. 2017; Rougeux et al. 2017). We found that a maximum
of seven mismatches for a 95 bp RAD tag offered the best compromise to correctly align both species and the outgroup to the _S. senegalensis_ genome. This was set to _bowtie 2_ options using
a minimum alignment score of --score-min = −42 needed for an alignment to be considered as valid, fixing the penalty of mismatch bases to MX = 6 and using default gap penalties. We then
used _pstacks_ to call variable positions under the bounded SNP model, setting the upper sequencing error rate to 2.5% and a minimum sequencing depth to 5× per stack. Homologous loci across
samples were merged based on their genomic position within scaffolds using _cstacks_ to construct a catalog of loci. Individual stacks were then matched against the catalog of loci with
_sstacks_ to determine genotypes. The module _rxstacks_ was ran with the --prune_haplo option to exclude poor-quality loci with multiple sequencing errors using a log-likelihood threshold of
−300 (determined empirically, Fig. S1). Individual genotypes were finally exported in the VCF format using the module _populations_. We then applied population-specific filters with
_VCFtools_ (Danecek et al. 2011) to remove SNPs showing significant deviation to Hardy–Weinberg equilibrium within at least one of the _S. senegalensis_ or _S. aegyptiaca_ locality samples
located outside the hybrid zone, using a _P_-value threshold of 0.01. Next, we excluded loci displaying more than 20% of missing genotypes in at least one locality. Over the 116,385
remaining SNPs, we randomly selected one single SNP for each pair of RAD loci associated with the same restriction site (i.e., within a distance less than 200 bp), in order to limit the
impact of linkage disequilibrium. Finally, we only retained loci with available sequence data in the outgroup species _S. solea_, which had to be fixed for the polymorphic site found in _S.
senegalensis_ and _S. aegyptiaca_. This resulted in a final dataset containing 10,758 independent SNPs that were specifically filtered to comply with the _δaδi_ analysis requirements. The
same SNPs were used in all subsequent analyses unless specified otherwise. GENETIC STRUCTURE AND HYBRIDIZATION Genetic variation within and among _S. senegalensis_ and _S. aegyptiaca_ was
characterized with a principal component analysis (PCA) performed with the R package _Adegenet_ (Jombart 2008; Jombart and Ahmed 2011). In order to document the possible existence of
population structure within each species and distinguish parental and admixed genotypes, we estimated individual admixture proportions from two to four differentiated genetic clusters that
were inferred from the data with no a priori information on individual membership, using _fastStructure_ (Raj et al. 2014) with 108 iterations. In addition, we estimated the proportion of
alleles with _S. aegyptiaca_ ancestry for each individual, using the _R_ package _Introgress_ (Gompert and Buerkle 2010) to estimate a Hybrid Index. We then searched specifically the
presence of early-generation hybrids using _NewHybrids_ (Anderson and Thompson 2002) with a subset of 296 diagnostic SNPs showing fixed differences between species. For each individual, we
estimated the membership probability to each of six categories of pedigree including pure parental species (P_sen_ and P_aeg_), first and second-generation hybrids (F1 and F2), and
first-generation backcrosses in each direction (BC_sen_ and BC_aeg_), using a total of 10,000 burn-in steps and 50,000 iterations. DEMO-GENETIC INFERENCE OF THE DIVERGENCE HISTORY We used a
modified version of the _δaδi_ program (Gutenkunst et al. 2009) that improves likelihood optimization using a simulated annealing (SA) procedure (Tine et al. 2014) in order to infer the
divergence history between _S. senegalensis_ and _S. aegyptiaca_. This method uses the joint allele frequency spectrum (JAFS) between two populations as summary statistics to characterize
divergence. The program _δaδi_ fits demographic divergence models to the observed data using a diffusion approximation of the JAFS, enabling a comparison of different alternative divergence
models in a composite likelihood framework. We used seven divergence models developed in a previous study (Tine et al. 2014) to determine whether and how gene flow has shaped genome
divergence between _S. senegalensis_ and _S. aegyptiaca_. The simplest model of strict isolation (SI) corresponds to an allopatric divergence scenario in which the ancestral population of
effective size _N_A splits into two derived populations of size _N_1 (_S. senegalensis_) and _N_2 (_S. aegyptiaca_) that evolve without exchanging genes for _T_s generations. We then
considered three models of divergence including gene flow, either during the entire divergence period (isolation with migration, IM), the beginning of divergence (AM), or the most recent
part of divergence (SC). In these models, migration occurs with potentially asymmetric rates (_m_12 and _m_21) that are shared across all loci in the genome. We also considered simple
extensions of the _IM_, _AM_, and _SC_ models that capture the effect of selection by accounting for heterogeneous migration rates among loci (_IM2m_, _AM2m_, and _SC2m_). These
semi-permeability models consider that two categories of loci, experiencing different effective migration rates, occur in proportions _P_ and 1−_P_ in the genome. The JAFS was obtained by
pooling the least introgressed populations from _S. senegalensis_ (Dakar, Cadiz, Annaba, and Mellah) for species 1 and _S. aegyptiaca_ (Kerkennah, El Biban, and Bardawil) for species 2 (Fig.
2), in order to avoid including admixed genotypes causing departures to the underlying population model. We also tested the robustness of our inferences with respect to the existence of a
subtle genetic structure within each species (Fig. 2a, b) by considering only Annaba and Mellah for _S. senegalensis_ (species 1) and Kerkennah and El Biban for _S. aegyptiaca_ (species 2).
We used _S. solea_ samples as an outgroup species to determine the most parsimonious ancestral allelic state for each SNP in order to generate an unfolded JAFS oriented with the derived
allele (Fig. 3a). The size of the JAFS was projected down to 40 sampled chromosomes per species to account for missing data. For each model, we estimated the parameter values that maximize
likelihood using two successive SA procedures before quasi-Newton (BFGS) optimization (Tine et al. 2014). Comparisons among models was made using the Akaike information criterion (AIC) to
account for variation in the number of parameters among models. A total of 20 independent runs were used for the optimization of each model. Parameter uncertainties were estimated from
non-parametric bootstrapped data using the Godambe information matrix as implemented in _δaδi_. We used 1000 bootstrapped datasets to estimate confidence intervals as the maximum likelihood
parameter value ±1.96 × SE. Estimated parameter values were converted into biologically meaningful quantities by taking into account our SNP filtering procedures following the method in
Rougeux et al. (2017). INFERRING THE PROBABILITY OF LOCUS INTROGRESSION UNDER THE BEST DIVERGENCE MODEL Because the JAFS-based inference of the divergence history does not provide an
assessment of introgression probability for each locus separately, we developed an approach to estimate the relative probability of each individual locus to be assigned to one of these two
categories: (i) loci which can readily introgress between species, and (ii) loci experiencing a highly reduced introgression rate due to selection against foreign alleles and linkage. This
probability was estimated using the best-fit model identified in the previous section, which was a SC model with variable introgression rates among loci (SC_2m_) (Fig. 3b). The SC2_m_ model
can be decomposed as a linear combination of two simple models describing gene flow in two different compartments of the genome (Fig. 3c). The best-fit SC_2m_ model estimated that only 5% of
the loci can still introgress between species with effective migration rates _m_1−2 and _m_2−1, whereas the remaining 95% of loci experience a highly reduced introgression rate with
effective migration rates _m_′1−2 and _m_′2−1. We used estimated model parameters to perform coalescent simulations with _msms_ (Ewing and Hermisson 2010) under the SC model (assuming theta
= 747.202; _N_1 = 0.818; _N_2 = 1.136; _T_S = 4.803; _T_SC = 0.081) using two different conditions for gene flow (assuming either _m_1−2 = 4.607 and _m_2−1 = 0.381, or _m_′1−2 = 0.057 and
_m_′2−1 = 0.153) to generate a _free introgression_ and a _reduced introgression_ rate dataset. One thousand JAFS with 40 sampled chromosomes per species were produced under each model. We
then averaged the number of derived alleles within entries across replicates to obtain a single JAFS for both the _free introgression_ (Y) and the _reduced introgression_ (Φ) model (Fig.
3c). Finally, we used the average number of SNPs per entry (_i_,_j_) in each JAFS to estimate the probability _P__i_,_j_ that a SNP with a derived allele count _i_ in species 1 and _j_ in
species 2 can be obtained under the _free introgression_ model: $$P_{i,j}{\mathrm{ = }}0.05{\mathrm{ \times \Upsilon }}_{i,j}/\left( {0.05 \times {\mathrm{\Upsilon }}_{i,j} + 0.95 \times
{\mathrm{\Phi }}_{i,j}} \right),$$ (1) where Y_i_,_j_ and Φ_i,j_ are the average number of SNPs predicted in the JAFS entry (_i_,_j_) under the _free introgression_ and _reduced
introgression_ model, respectively. Every SNP from the real dataset was finally associated to the introgression probability _P__i,j_ given by Eq. (1) based on its corresponding entry in the
JAFS. GENOMIC CLINES The Bayesian Genomic Cline program _BGC_ (Gompert and Buerkle 2012) was used to quantify individual locus introgression relative to genome-wide introgression. _BGC_
describes the probability of locus-specific ancestry from one parental species given the genome-wide hybrid index. The _BGC_ model considers two principal parameters, called _α_ and _β_,
which describe locus-specific introgression based on ancestry. Parameter _α_ quantifies the change in probability of ancestry relative to a null expectation based on genome-wide hybrid
index, that is, the direction of introgression. A positive value of _α_ reflects an increase in the probability of ancestry from species 1 (introgression into _S. aegyptiaca_), whereas a
negative value of _α_ reflects an increase in the probability of ancestry from species 2 (introgression into _S. senegalensis_). Parameter _β_ describes the rate of transition in the
probability of ancestry from parental population 1 to parental population 2 as a function of hybrid index, that is, the amount of introgression. Positive _β_ values thus denote a restricted
amount of introgression, whereas negative _β_ values indicate a greater introgression rate compared to the genome-wide average. We ran _BGC_ under the genotype-uncertainty model, assuming a
sequence error probability of 0.0001 and using information from the data to initialize ancestry and hybrid index. Two MCMC chains each made of 150,000 steps were ran, recording every 20th
value. Cline parameter quantiles were calculated to designate outlier loci with respect to parameters _α_ and _β_ based on the assumption that the genome-wide distributions of locus-specific
cline parameters are both centered on zero. Therefore, outlier loci are markers with extreme patterns of introgression relative to the remainder of the genome. A locus was considered as an
_α_ outlier if its posterior estimate of _α_ was not contained in the interval bounded by the 0.025 and 0.975 quantiles of \({\cal N}(0,\tau _\alpha )\) (likewise for _β_) (Gompert and
Buerkle 2011; Gompert et al. 2013). GEOGRAPHIC CLINES The geographic cline analysis was carried out in order to link allele frequencies at individual loci with geographic position along a
transect spanning the hybrid zone. We used the R package _HZAR_ (Derryberry et al. 2014) that fits allele frequency data to classic equilibrium models of geographic clines (Szymura and
Barton 1986) using the MCMC algorithm. Instead of searching for the best model separately for each cline, we used the full model that fits cline center, width, and independent introgression
tails using estimated values for minimum (_p_min) and maximum (_p_max) allele frequencies. In this way, we avoided the potentially confounding effects of fitting different models for the
comparison of cline center and slope parameters across loci. RESULTS A total of 833.3 million raw reads were obtained, 89.1% of which were retained after demultiplexing and quality filtering
for reference mapping against the _S. senegalensis_ draft genome (average number of reads per individual: 3,906,809, s.d.=1,878,685). Individual genotype calling in _Stacks_ produced a raw
VCF file containing 174,490 SNPs, from which we retained 10,758 oriented SNPs (using _S. solea_ as an outgroup to identify ancestral states) after controlling for linkage and filtering for
quality. The mean sequencing depth per locus per individual was superior to 100× (Fig. S2), and the mean genotype missing rate was 1.3% (s.d. = 2.6%) per individual and 1.4% (s.d. = 1.8%)
per locus. The final VCF containing 10,758 SNPs was used for all downstream analyses except _NewHybrids_. GENETIC STRUCTURE, HYBRIDIZATION, AND INTROGRESSION The PCA clearly separated _S.
senegalensis_ from _S. aegyptiaca_ samples along the first PC axis (PC1), which explained 74.2% of the total genotypic variance (Fig. 2a). _S. senegalensis_ samples were organized along that
axis according to their geographical proximity from the contact zone, following a gradient of genetic similarity to _S. aegyptiaca_ increasing from Dakar to Bizerte lagoon. The two species
were found to coexist only in Bizerte lagoon, with four _S. aegyptiaca_ individuals being found among a majority of _S. senegalensis_ genotypes. The second principal component revealed a
weak signal of differentiation (PC2, 0.73% of associated variance) separating Bardawil lagoon from other samples within _S. aegyptiaca_. Likewise, the third principal component (PC3, 0.66%
of genotypic variance) captured a subtle differentiation signal between the Atlantic samples from Dakar and Cadiz and the Mediterranean _S. senegalensis_ samples (Fig. 2b). The
_fastStructure_ analysis (Fig. 2c) confirmed the separation of the two species into different genetic clusters and their coexistence in Bizerte lagoon, as detected from the PCA. However, it
failed to detect any further genetic subdivision (Fig. S3), confirming that the signal of genetic structure within each species is at most very small. The finding of intermediate admixture
proportions in some individuals revealed signs of introgressive hybridization between _S. senegalensis_ and _S. aegyptiaca_ around the contact zone. The relationship between individual
hybrid index and interspecific heterozygosity represented in a triangle plot illustrated well the absence of F1 and F2 hybrids in our dataset, and the presence of a few backcrosses together
with several introgressed genotypes (i.e., late-generation backcrosses) (Fig. 2d). This was confirmed by the detection of three first generation backcrosses by _NewHybrids_ (detected with an
assignment probability of 1), one in the direction of _S. senegalensis_ (in Bizerte lagoon) and two in the opposite direction (in Gulf of Tunis), plus two unassigned individuals likely
representing later-generation backcrosses (Fig. S4). Therefore, our results provide evidence for contemporary introgressive hybridization between the two sole species. DEMO-GENETIC HISTORY
OF DIVERGENCE The _δaδi_ analysis showed that the SC model with varying introgression rates along the genome best explained the observed JAFS (SC2_m_, delta AIC with the second ranked model
>25). In comparison, the six other models had a significantly lower performance for different reasons. While the SI model could explain SNPs occupying the outer frame of the JAFS, it
could not predict at the same time the presence of loci in the more inner part of the spectrum. The IM, AM, and SC models assuming genome-wide homogeneous migration rates better predicted
loci occupying the central part of the JASF. However, they underestimated the density of private and highly differentiated SNPs. Finally, the three models including heterogeneous migration
rates along the genome (IM2_m_, AM2_m_, and SC2_m_) yielded significantly improved fits. Among them, only the SC2_m_ model provided a good prediction for both a high density of highly
differentiated SNPs between the two species and the presence of SNPs toward the central part of the spectrum. Model selection was not sensible to the potential effect of unaccounted
structure within each species (Table S1), consistent with the very weak signals of within-species differentiation detected in PCA analysis. Using the best-fit obtained for the SC2_m_ model
over 20 independent runs to get estimates of model parameters and confidence intervals, we found that the duration of the isolation period between _S. senegalensis_ and _S. aegyptiaca_ was
about 60 times longer than the duration of the SC. A large proportion of the genome (~95%) was associated with relatively small effective migration rates and limited gene flow corresponding
to less than one effective migrant per generation in both directions (_N_1_m_′12 = 0.047, _N_2_m′_21 = 0.175). By contrast, the effective migration rate was more elevated in the remaining
small fraction of the genome (~5%), especially in the direction from _S. senegalensis_ to _S. aegyptiaca_ where introgression was found 80 times higher than elsewhere in the genome.
Introgression was found 12 times lower in direction of _S. senegalensis_, although still more than twice as high than in the remainder (95%) of the genome (Table 1). GENOMIC CLINES The
genomic cline analysis performed with the _BGC_ program revealed different behaviors among the 10,758 SNPs with respect to the cline parameters _α_ and _β_. Considering the locus-wise
ancestry shift parameter _α_, we observed that 48% of the loci have an excess of _S. senegalensis_ ancestry (negative _α_), whereas only 29% of them have an excess of _S. aegyptiaca_
ancestry (positive _α_). The quantile method for outlier detection confirmed that a higher proportion of loci was characterized by an excess of _S. senegalensis_ ancestry (43% of negative
_α_ outliers) compared to _S. aegyptiaca_ ancestry (17% of positive _α_ outliers). By contrast, the locus-wise slope parameter _β_ depicting the rate of transition between species was
symmetrically distributed between negative and positive values, with equal proportions of loci showing a decreased (48% of positive _β_) and increased (51% of negative _β_) introgression
rates. The proportion of loci showing significant deviation compared to the genomic average amounted to 52%, and were equally distributed among negative (26%) and positive (26%) _β_
outliers. GEOGRAPHIC CLINES The mean position of cline center (_C_) calculated over all fitted clines was located 12 km to the east of Bizerte lagoon (Fig. 4). Most individual locus clines
tended to co-localize at this position, especially for the steepest clines whose centers localized essentially 10 km or less around the center of the hybrid zone. In general, loci whose
individual centers did not coincide with the central part of the hybrid zone harbored less steep clines, either because they correspond to rare variants with low information content (i.e.,
that do not form a cline) or due to softer selection at these loci which are uncoupled from the hybrid zone (Fig. 4). LINKS BETWEEN DIFFERENT APPROACHES Linear regression models were used to
investigate whether different analyses of individual locus introgression that use different aspects of the data tend to produce similar results. First, we evaluated the extent to which an
excess of ancestry from a given species relates to a spatial shift of cline center into the other species range. The genomic cline parameter _α_ showed a significant positive correlation
with the geographic cline center _C_ on both sides of the hybrid zone (Fig. 5). The correlation was, however, much stronger in the _S. aegyptiaca_ side (_R_²c−α = 0.274, _P_ < 10−10)
compared to the _S. senegalensis_ side where it was barely detectable (_R_²c−α = 0.023, _P_ < 10−10). We then tested whether the loci exhibiting the most abrupt transition in ancestry
between the two species (i.e., a decreased introgression rate) also tended to display the steepest geographic clines. To validate this prediction, we focused on the 26% of positive _β_
outliers detected with _BGC_, and compared _β_ values with the slope parameter _S_ of the geographic clines. We found a significantly positive correlation (_R_²S−β = 0.27, _P_ < 10−10)
between the two parameters, confirming that loci with a low introgression rate also tend to display steeper geographic clines (Fig. 6). Moreover, we tried to connect genomic and geographic
cline parameters with our estimated probability that individual loci belong to the small fraction of the genome showing the highest introgression rate. As expected, geographic cline slope
was significantly negatively correlated with the inferred probability of introgression (_R_²S−Proba = 0.04, _P_ < 10−10, Fig. S5). Therefore, private SNPs located on the outer frame of
the JAFS, for which we inferred a small probability of introgression, were usually associated with steep cline slopes. By contrast, loci occupying the most central part of the JAFS, which
were assigned the highest introgression probabilities, were also characterized by shallower geographic clines. Finally, when restricting the analysis to the 5% of loci with the highest
introgression probability, we show that a majority of their geographic cline centers are located outside the contact zone, with a spatial shift more pronounced in the _S. aegyptiaca_
direction (Fig. 7a). Similarly, the distribution of the genomic cline parameter _α_ also showed a deficit of values around zero, and a majority of loci with an excess of _S. senegalensis_
ancestry, in keeping with the general asymmetry of the exchanges between the two species (Fig. 7b). DISCUSSION DIVERGENCE HISTORY AND SEMI-PERMEABILITY TO GENE FLOW Our results using
high-throughput genotyping confirmed previous observations, based on a limited number of nuclear markers, that the flatfish _S. senegalensis_ and _S. aegyptiaca_ are genetically divergent
sibling species, which are still exchanging genes across their natural hybrid zone near Bizerte lagoon (She et al. 1987; Ouanes et al. 2011; Souissi et al. 2017). However, the proportion of
hybrid genotypes detected here in the contact zone (i.e., 3 early generation hybrids among 54 individuals) is much lower than established in previous studies. Although this could be due to
large temporal variations in the rate of hybridization, the most plausible hypothesis is that real hybrids (e.g., F1, F2, and first generations backcrosses) are more easily distinguished
from introgressed genotypes using a high number of diagnostic markers (Pujolar et al. 2014; Jeffery et al. 2017), whereas earlier studies have used principally non-diagnostic markers. Our
results thus support the existence of an abrupt geographic transition between the two species across their contact zone, where predominantly parental genotypes occur in sympatry with an
overall low frequency of hybrids. These observations are consistent with the tension zone model (Barton and Hewitt 1985), in which the hybrid zone is maintained by a balance between the
influx of parental genotypes from outside the zone and the counter-selection of hybrid genotypes inside the zone. This interpretation is also in keeping with the finding of transgressive
body shape variation attributed to a reduced condition index of admixed genotypes from the contact zone (Souissi et al. 2017). The regions located outside the contact zone, which contain
only parental genotypes, provided relevant information to infer the demo-genetic history of divergence between these two species. Gene exchange between _S. senegalensis_ and _S. aegyptiaca_
is best explained by a SC model with heterogeneous rates of introgression along the genome. Similar divergence histories have already been found between geographical lineages or ecotypes of
the same species, like in the European sea bass and anchovy (Tine et al. 2014; Le Moan et al. 2016). Using the same approach as implemented here, these studies could determine that a minor
but significant fraction of the genome (i.e., 20–35%) does not neutrally introgress between hybridizing lineages/ecotypes, thus providing a quantitative assessment of the degree of
semi-permeability to gene flow. In the present case, by contrast, we found that the great majority of the genome (approximately 95%) experiences a highly reduced effective migration rate
(i.e., <1 migrant per generation) between species. Moreover, the inferred divergence period estimated using a mutation rate of 10−8 per bp per year and a generation time of 3–5 years
corresponds to a separation time of _ca_. 1.1 to 1.8 Myrs, which together with the mitochondrial sequence divergence of 2%, indicates a relatively ancient speciation event. This timing of
divergence is much older than the approximately 300 Kyrs that were inferred between glacial lineages in anchovy and sea bass (Tine et al. 2014; Le Moan et al. 2016). Therefore, our results
are consistent with the prediction that more anciently diverged species should display stronger reproductive isolation due to the establishment of more genetic barriers (Orr 1995; Moyle and
Nakazato 2010; Roux et al. 2016), which is reflected here by a lower level of permeability to gene flow compared to sea bass and anchovy. Another aspect of this low miscibility of the two
genomes was captured by individual locus cline parameters. Consistent with the view that stepped clines generate a strong barrier to gene flow (Szymura and Barton 1986), we found a high
proportion of loci combining extremely low introgression rates (i.e., positive _β_ outliers) and steep geographic clines (i.e., cline width below the average) with centers colocalized in the
central part of the hybrid zone. Because this type of situation generates strong linkage disequilibrium, each locus is expected to receive, in addition to its own selective coefficient, the
effect of selection on other loci (Barton 1983; Kruuk et al. 1999). This makes the genome acting almost as a single underdominant locus causing a global reduction in gene flow, which
sometimes is referred to as a “congealed genome” (Turner 1967; Bierne et al. 2011; Gompert et al. 2012b) relatively to its very low capacity to incorporate foreign genes. In contrast to this
genome-wide reduction in gene flow, we also found support for a small proportion of genomic regions with higher-than-average introgression rates between species. MULTIPLE APPROACHES TO
DETECT INCREASED INTROGRESSION RATES Differential introgression among loci was evidenced by combining several approaches that capture different but complementary aspects of the data. For
instance, genomic and geographic cline methods both rely on the sampling of admixed genotypes from the hybrid zone, whereas the modeling approach based on the JAFS relies of the joint
distribution of allele frequencies between parental populations located outside of the hybrid zone. By combining these methods, we found that the 5% of loci with higher-than-average
introgression rates identified with the JASF approach generally display shifts in both geographic and genomic cline centers, as illustrated by their tendency to have non-zero cline center
and _α_ parameter values (Fig. 7). Therefore, our method to detect increased introgression while accounting for the demographic divergence history identifies genomic regions that also depart
from the genome average in those places where the two species meet and admix. Discordant clines were not symmetrically distributed between the _S. senegalensis_ and _S. aegyptiaca_ side of
the hybrid zone. The majority of the highly introgressed loci were shifted into the _S. aegyptiaca_ geographic range. For these loci, the excess of _S. senegalensis_ ancestry (measured by
the genomic cline parameter _α_) was positively related to the extent of the spatial shift of the cline center into the _S. aegyptiaca_ territory. This correlation was much stronger than
previously observed in a bird hybrid zone study (Grossen et al. 2016), possibly due to a stronger variance in cline shift in the _Solea_ system because of hybrid zone movement (see below).
By contrast, much fewer loci were found with increased introgression rates in the opposite direction. Such loci generally displayed cline centers shifted away from the contact zone, probably
because the Atlantic samples of _S. senegalensis_ are introgressed to a lesser extent, if any, than those inside the Mediterranean as evidenced by their position along axis one of the PCA
(Fig. 2b). The spatial shift in cline center was in this case only weakly positively correlated with the excess of _S. aegyptiaca_ ancestry. This could be expected, however, since the
genomic cline method has limited power to infer cline parameters outside the range of observed hybrid indexes in admixed genotypes (Gompert et al. 2012a), which in our case were biased in
favor of _S. aegyptiaca_ ancestry. DIFFERENTIAL INTROGRESSION PATTERNS AND THE DYNAMICS OF THE HYBRID ZONE The inferred history of divergence between _S. senegalensis_ and _S. aegyptiaca_
places the beginning of gene flow _ca_. 18 to 30 Kyrs ago, supporting a scenario of SC at the end of the last glacial period. Therefore, the hybrid zone is probably sufficiently old for the
dynamics of introgression to be well established. This supports the idea that reproductive isolation between _S. senegalensis_ and _S. aegyptiaca_ was strong enough to prevent genetic
homogenization throughout most of the genome during this period. At the same time, the small fraction of introgressing loci that were still able to cross the species barrier indicate the
existence of genomic regions unlinked to reproductive isolation loci, either due to a local lack of barrier loci and/or to a high local recombination rate (Roux et al. 2013). Whether
prezygotic or postzygotic effects could explain the observed asymmetric introgression under the hypothesis of a stable hybrid zone remains to be fully addressed. Alternatively, the
geographic pattern of asymmetrical introgression evidenced here could reveal a movement of the hybrid zone (Buggs 2007), especially if the markers concerned are randomly spread across the
genome (Wielstra et al. 2017). In the absence of a well-assembled reference genome, the genomic distribution of introgressing loci could not be addressed. However, a northward movement of
the hybrid zone has already been proposed based on a temporal trend of increasing abundance of _S. aegyptiaca_ in Bizerte lagoon (Ouanes et al. 2011). Therefore, our results probably reflect
a recent or ongoing shift of the hybrid zone, with _S. aegyptiaca_ incorporating the small fraction of compatible genes from _S. senegalensis_ as it moves northward in its territory,
leaving behind a tail of neutral or quasi-neutral introgressed alleles. Similar signatures have already been reported in other sister species such as in house mice (Wang et al. 2011),
rabbits (Carneiro et al. 2013), salamanders (Visser et al. 2017), toads (Arntzen et al. 2017), newts (Wielstra et al. 2017), and chickadees (Taylor et al. 2014). The possible causes of
hybrid zone movement include the tracking of environmental changes, fitness differences among individuals from the two species, or gradients of population density (Barton and Hewitt 1985;
Buggs 2007; Taylor et al. 2015; Gompert et al. 2017). Future studies will have to establish which of these hypotheses accounts for the asymmetric introgression pattern in soles. Opposite to
the preferential direction of introgression, we also found a few loci within the _S. senegalensis_ genetic background whose geographic center was apparently shifted far away from the contact
zone. This pattern was also captured by the first and third PCA axes, along which we observed a slight differentiation between the _S. senegalensis_ samples from Dakar and Cadiz on one side
and Annaba on the other, which seems to be congruent with increased introgression in Mediterranean compared to Atlantic _S. senegalensis_ samples. Two non-mutually exclusive explanations
can be put forward to account for this difference. Strong barriers to gene flow only have a delaying effect on the dynamics of introgression (Barton 1979). If the barrier is strong enough,
many generations may be required for advantageous or simply neutral alleles to extricate themselves from the hybrid zone, depending on the balance between the selective coefficient of the
focal alleles and that of the deleterious genes contained in their chromosomal vicinity. Hence, spatial allele frequency patterns may appear even well after SC, and since the entrance of the
Mediterranean Sea is a well-known barrier to dispersal for many marine organisms (Patarnello et al. 2007), the wave of advance of introgression clines may be delayed as they travel toward
Dakar to the south of _S. senegalensis_’ range. Nevertheless, advantageous alleles are expected to race ahead neutral ones once they are freed from their background, a possibility would thus
be that some of these loci exhibiting a shifted center could be related to an adaptive introgression of _S. aegyptiaca_ alleles into the _S. senegalensis_ background. Evidence for adaptive
alleles spreading between species has been found in recent hybrid zones induced by human introductions (Fitzpatrick et al. 2010), but should be difficult to observe in historical hybrid
zones where advantageous alleles have had ample time to spread (Hewitt 1988). Alternatively, these clines may correspond to alleles providing a local advantage to the _S. senegalensis_
populations in the Mediterranean environment but not in the Atlantic. The hypothesis of adaptive introgression needs to be scrutinized in more details by focusing on the chromosomal
signature of differentiation around the putative selected locus (Bierne 2010). CONCLUSION To conclude, our combination of analytical approaches provides new insights into the genomic
architecture and the dynamics of gene flow between two divergent but still interacting parapatric species. Despite a relatively modest geographic coverage and the scarcity of available
admixed genotypes in the tension zone, the genome-wide analyses taking into account the inferred history of divergence provided an efficient way to detect loci with deviant introgression
behaviors. This is complementary to classical approaches based on genomic and geographic cline analyses. Our results bring new support for the tension zone model against a simple coexistence
in sympatry. We show that differential gene flow has shaped genetic divergence across the tension zone, although most loci behave as if they were sitting on a congealed genome, rendering
very unlikely the future remixing of the two genes pools via the creation of a hybrid swarm. Nevertheless, a few genes seem able to escape counter-selection, possibly due to different
underlying processes. The first involves a possible movement of the zone, in which a shift of species range boundary leaves a tail of neutral introgressed alleles behind the front of the
invading species. The second is possibly related to the existence of a physical barrier to gene flow near Gibraltar and/or the spread of globally or locally adaptive alleles into the range
of the invaded species. Our results thus provide a snapshot on the genetic outcome of evolutionary processes potentially involved when divergent gene pools come back into contact after a
long period of geographical isolation. DATA ARCHIVING Demultiplexed RAD sequencing read data (fastq files) are available on NCBI under BioProject Accession: PRJNA443143. VCF files and input
files for _dadi_ are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.h508g38. REFERENCES * Anderson EC, Thompson EA (2002) A model-based method for identifying
species hybrids using multilocus genetic data. Genetics 160:1217–1229 CAS PubMed PubMed Central Google Scholar * Arntzen JW, de Vries W, Canestrelli D, Martínez-Solano I (2017) Hybrid
zone formation and contrasting outcomes of secondary contact over transects in common toads. Mol Ecol 26:5663–5675 Article CAS Google Scholar * Baird NA, Etter PD, Atwood TS, Currey MC,
Shiver AL, Lewis ZA et al. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PloS ONE 3:e3376 Article Google Scholar * Barton NH (1979) Gene flow past a cline.
Heredity 43:333–339 Article Google Scholar * Barton NH (1983) Multilocus clines. Evolution 37:454–471 Article CAS Google Scholar * Barton N, Bengtsson BO (1986) The barrier to genetic
exchange between hybridising populations. Heredity 57:357–376 Article Google Scholar * Barton NH, Gale KS (1993) Genetic analysis of hybrid zones. In: Harrison RG (ed.) Hybrid Zones and
the Evolutionary Process. Oxford University Press, pp. 13–45 * Barton NH, Hewitt GM (1985) Analysis of hybrid zones. Annu Rev Ecol Syst 16:113–148 Article Google Scholar * Bierne N (2010)
The distinctive footprints of local hitchhiking in a varied environment and global hitchhiking in a subdivided population. Evolution 64:3254–3272 Article CAS Google Scholar * Bierne N,
Borsa P, Daguin C, Jollivet D, Viard F, Bonhomme F et al. (2003) Introgression patterns in the mosaic hybrid zone between _Mytilus edulis_ and _M. galloprovincialis_. Mol Ecol 12:447–461
Article CAS Google Scholar * Bierne N, Welch J, Loire E, Bonhomme F, David P (2011) The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20:2044–2072
Article Google Scholar * Buggs RJA (2007) Empirical study of hybrid zone movement. Heredity 99:301 Article CAS Google Scholar * Carneiro M, Baird SJE, Afonso S, Ramirez E, Tarroso P,
Teotonio H et al. (2013) Steep clines within a highly permeable genome across a hybrid zone between two subspecies of the European rabbit. Mol Ecol 22:2511–2525 Article CAS Google Scholar
* Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140 Article Google Scholar * Coyne JA, Orr HA
(2004) Speciation. Sinauer Associates, Inc., Sunderland, MA Google Scholar * Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al. (2011) The variant call format and
VCFtools. Bioinformatics 27:2156–2158 Article CAS Google Scholar * Derryberry EP, Derryberry GE, Maley JM, Brumfield RT (2014) HZAR: hybrid zone analysis using an R software package. Mol
Ecol Resour 14:652–663 Article Google Scholar * Dobzhansky T (1937) Genetic nature of species differences. Am Nat 71:404–420 Article Google Scholar * Endler JA (1977) Geographic
variation, speciation, and clines. Princeton University Press * Ewing G, Hermisson J (2010) MSMS: a coalescent simulation program including recombination, demographic structure and selection
at a single locus. Bioinformatics 26:2064–2065 Article CAS Google Scholar * Fitzpatrick BM (2013) Alternative forms for genomic clines. Ecol Evol 3:1951–1966 Article Google Scholar *
Fitzpatrick BM, Johnson JR, Kump DK, Smith JJ, Voss SR, Shaffer HB (2010) Rapid spread of invasive genes into a threatened native species. Proc Natl Acad Sci USA 107:3606–3610 Article CAS
Google Scholar * Gompert Z, Buerkle C (2009) A powerful regression-based method for admixture mapping of isolation across the genome of hybrids. Mol Ecol 18:1207–1224 Article Google
Scholar * Gompert Z, Buerkle CA (2010) introgress: a software package for mapping components of isolation in hybrids. Mol Ecol Resour 10:378–384 Article CAS Google Scholar * Gompert Z,
Buerkle CA (2011) Bayesian estimation of genomic clines. Mol Ecol 20:2111–2127 Article Google Scholar * Gompert Z, Buerkle CA (2012) bgc: software for Bayesian estimation of genomic
clines. Mol Ecol Resour 12:1168–1176 Article CAS Google Scholar * Gompert Z, Lucas LK, Nice CC, Buerkle CA (2013) Genome divergence and the genetic architecture of barriers to gene flow
between Lycaeides Idas and L-Melissa. Evolution 67:2498–2514 Article Google Scholar * Gompert Z, Lucas LK, Nice CC, Fordyce JA, Forister ML, Buerkle CA (2012a) Genomic regions with a
history of divergent selection affect fitness of hybrids between two butterfly species. Evolution 66:2167–2181 Article Google Scholar * Gompert Z, Mandeville EG, Buerkle CA (2017) Analysis
of population genomic data from hybrid zones. Annu Rev Ecol Evol Syst. 48:207–229 Article Google Scholar * Gompert Z, Parchman TL, Buerkle CA (2012b) Genomics of isolation in hybrids
Philos Trans R Soc Lond B Biol Sci 367:439–450 Article Google Scholar * Grossen C, Seneviratne SS, Croll D, Irwin DE (2016) Strong reproductive isolation and narrow genomic tracts of
differentiation among three woodpecker species in secondary contact. Mol Ecol 25:4247–4266 Article CAS Google Scholar * Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD (2009)
Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet 5:e1000695 Article Google Scholar * Harrison RG (1990) Hybrid zones:
windows on evolutionary process. Oxf Surv Evol Biol 7:69–128 Google Scholar * Harrison RG, Larson EL (2016) Heterogeneous genome divergence, differential introgression, and the origin and
structure of hybrid zones. Mol Ecol 25:2454–2466 Article Google Scholar * Harrison RG and Rand DM (1989) Mosaic hybrid zones and the nature of species boundaries. In: Otte D and Endler JA
(eds) Speciation and its Consequences.Sinauer, Sunderland, MA, pp. 111–133 * Hewitt GM (1988) Hybrid zones-natural laboratories for evolutionary studies. Trends Ecol Evol 3:158–167 Article
CAS Google Scholar * Jeffery NW, DiBacco C, Van Wyngaarden M, Hamilton LC, Stanley RRE, Bernier R et al. (2017) RAD sequencing reveals genomewide divergence between independent invasions
of the European green crab (Carcinus maenas) in the Northwest Atlantic. Ecol Evol 7:2513–2524 Article Google Scholar * Jombart T (2008) adegenet: a R package for the multivariate analysis
of genetic markers. Bioinformatics 24:1403–1405 Article CAS Google Scholar * Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics
27:3070–3071 Article CAS Google Scholar * Kruuk LE, Baird SJ, Gale KS, Barton NH (1999) A comparison of multilocus clines maintained by environmental adaptation or by selection against
hybrids. Genetics 153:1959–1971 CAS PubMed PubMed Central Google Scholar * Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359 Article CAS
Google Scholar * Larson EL, Andrés JA, Bogdanowicz SM, Harrison RG (2013) Differential introgression in a mosaic hybrid zone reveals candidate barrier genes. Evolution 67:3653–3661 Article
CAS Google Scholar * Manchado M, Planas, JV, Cousin X, Rebordinos L, Claros MG (2016) Current status in other finfishspecies. In: MacKenzie S. and Jentoft S. (eds) Genomics in
Aquaculture. Academic Press * Le Moan A, Gagnaire P-A, Bonhomme F (2016) Parallel genetic divergence among coastal–marine ecotype pairs of European anchovy explained by differential
introgression after secondary contact. Mol Ecol 25:3187–3202 Article Google Scholar * Martinsen GD, Whitham TG, Turek RJ, Keim P (2001) Hybrid populations selectively filter gene
introgression between species. Evolution 55:1325–1335 Article CAS Google Scholar * Mayr, E. (1942) Systematics and the Origin of Species. New York, Columbia Univ. Press * Moyle LC,
Nakazato T (2010) Hybrid incompatibility “snowballs” between Solanum species. Science 329:1521–1523 Article CAS Google Scholar * Orr HA (1995) The population genetics of speciation: the
evolution of hybrid incompatibilities. Genetics 139:1805–1813 CAS PubMed PubMed Central Google Scholar * Ouanes K, Bahri-Sfar L, Ben Hassine OK, Bonhomme F (2011) Expanding hybrid zone
between _Solea aegyptiaca_ and _Solea senegalensis_: genetic evidence over two decades. Mol Ecol 20:1717–1728 Article CAS Google Scholar * Patarnello T, Volckaert FA, Castilho R (2007)
Pillars of Hercules: is the Atlantic–Mediterranean transition a phylogeographical break? Mol Ecol 16:4426–4444 Article Google Scholar * Payseur BA (2010) Using differential introgression
in hybrid zones to identify genomic regions involved in speciation. Mol Ecol Resour 10:806–820 Article Google Scholar * Payseur BA, Rieseberg LH (2016) A genomic perspective on
hybridization and speciation. Mol Ecol 25:2337–2360 Article CAS Google Scholar * Porter AH, Wenger R, Geiger H, Scholl A, Shapiro AM (1997) The Pontia daplidice-edusa hybrid zone in
northwestern Italy. Evolution 51:1561–1573 PubMed Google Scholar * Pujolar JM, Jacobsen MW, Als TD, Frydenberg J, Magnussen E, Jónsson B et al. (2014) Assessing patterns of hybridization
between North Atlantic eels using diagnostic single-nucleotide polymorphisms. Heredity 112:627 Article CAS Google Scholar * Raj A, Stephens M, Pritchard JK (2014) fastSTRUCTURE:
variational inference of population structure in large SNP data sets. Genetics 197:573–589 Article Google Scholar * Rougemont Q, Gagnaire P-A, Perrier C, Genthon C, Besnard A-L, Launey S
et al. (2017) Inferring the demographic history underlying parallel genomic divergence among pairs of parasitic and nonparasitic lamprey ecotypes. Mol Ecol 26:142–162 Article CAS Google
Scholar * Rougeux C, Bernatchez L, Gagnaire P-A (2017) Modeling the multiple facets of speciation-with-gene-flow toward inferring the divergence history of Lake Whitefish species pairs
(_Coregonus clupeaformis_). Genome Biol Evol 9:2057–2074 Article Google Scholar * Roux C, Fraisse C, Romiguier J, Anciaux Y, Galtier N, Bierne N (2016) Shedding light on the grey zone of
speciation along a continuum of genomic divergence. PLoS Biol 14:e2000234 Article Google Scholar * Roux C, Tsagkogeorga G, Bierne N, Galtier N (2013) Crossing the species barrier: genomic
hotspots of introgression between two highly divergent Ciona intestinalis species. Mol Biol Evol 30:1574–1587 Article CAS Google Scholar * She JX, Autem M, Kotulas G, Pasteur N, Bonhomme
F (1987) Multivariate-analysis of genetic exchanges between _Solea aegyptiaca_ and _Solea senegalensis_ (Teleosts, Soleidae). Biol J Linn Soc 32:357–371 Article Google Scholar * Souissi A,
Gagnaire PA, Bonhomme F, Bahri-Sfar L (2017) Introgressive hybridization and morphological transgression in the contact zone between two Mediterranean Solea species. Ecol Evol 7:1394–1402
Article Google Scholar * Sousa VC, Carneiro M, Ferrand N, Hey J (2013) Identifying loci under selection against gene flow in isolation-with-migration models. Genetics 194:211–233 Article
CAS Google Scholar * Szymura JM, Barton NH (1986) Genetic analysis of a hybrid zone between the fire-bellied toads, Bombina bombina and B. variegata, near Cracow in southern Poland.
Evolution 40:1141–1159 PubMed Google Scholar * Taylor SA, Curry RL, White TA, Ferretti V, Lovette I (2014) Spatiotemporally consistent genomic signatures of reproductive isolation in a
moving hybrid zone. Evolution 68:3066–3081 Article Google Scholar * Taylor SA, Larson EL, Harrison RG (2015) Hybrid zones: windows on climate change. Trends Ecol Evol 30:398–406 Article
Google Scholar * Teeter KC, Payseur BA, Harris LW, Bakewell MA, Thibodeau LM, O’Brien JE et al. (2008) Genome-wide patterns of gene flow across a house mouse hybrid zone. Genome Res
18:67–76 Article CAS Google Scholar * Tine M, Kuhl H, Gagnaire PA, Louro B, Desmarais E, Martins RST et al. (2014) European sea bass genome and its variation provide insights into
adaptation to euryhalinity and speciation. Nat Commun 5:5770 Article CAS Google Scholar * Turner JR (1967) Why does the genotype not congeal? Evolution 21:645–656 Article Google Scholar
* Visser M, de Leeuw M, Zuiderwijk A, Arntzen JW (2017) Stabilization of a salamander moving hybrid zone. Ecol Evol 7:689–696 Article Google Scholar * Wang LY, Luzynski K, Pool JE,
Janousek V, Dufkova P, Vyskocilova MM et al. (2011) Measures of linkage disequilibrium among neighbouring SNPs indicate asymmetries across the house mouse hybrid zone. Mol Ecol 20:2985–3000
Article Google Scholar * Wielstra B, Burke T, Butlin RK, Avci A, Üzüm N, Bozkurt E (2017) A genomic footprint of hybrid zone movement in crested newts Evol Lett 1:93–101 Article Google
Scholar Download references ACKNOWLEDGEMENTS This work was conducted during the Ph.D. thesis of AS with the financial support of program PHC Maghreb No. XE27961 from the French Ministère
des Affaires Etrangères to FB and Ministère de l’Enseignement Supérieur et de la Recherche to LB-S. The authors are indebted to M-T Augé for skillful technical assistance and thank Nicolas
Bierne for insightful discussions. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Université de Montpellier, Place Eugène Bataillon, 34095, Montpellier, France Ahmed Souissi, François
Bonhomme & Pierre-Alexandre Gagnaire * CNRS—Institut des Sciences de l’Evolution, UMR5554 UM-CNRS-IRD-EPHE, Station Méditerranéenne de l’Environnement Littoral, 34200, Sète, France Ahmed
Souissi, François Bonhomme & Pierre-Alexandre Gagnaire * Faculté des Sciences de Tunis UR11ES08 Biologie intégrative et écologie évolutive et fonctionnelle des milieux aquatiques,
Université de Tunis El Manar, 2092, Tunis, Tunisia Ahmed Souissi & Lilia Bahri-Sfar * IFAPA Centro El Toruño, Junta de Andalucía, Camino Tiro Pichón s/n, 11500, El Puerto de Santa María,
Cádiz, Spain Manuel Manchado Authors * Ahmed Souissi View author publications You can also search for this author inPubMed Google Scholar * François Bonhomme View author publications You
can also search for this author inPubMed Google Scholar * Manuel Manchado View author publications You can also search for this author inPubMed Google Scholar * Lilia Bahri-Sfar View author
publications You can also search for this author inPubMed Google Scholar * Pierre-Alexandre Gagnaire View author publications You can also search for this author inPubMed Google Scholar
CORRESPONDING AUTHOR Correspondence to Ahmed Souissi. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ELECTRONIC SUPPLEMENTARY MATERIAL
SUPPLEMENTARY MATERIAL RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Souissi, A., Bonhomme, F., Manchado, M. _et al._ Genomic and geographic footprints
of differential introgression between two divergent fish species (_Solea_ spp.). _Heredity_ 121, 579–593 (2018). https://doi.org/10.1038/s41437-018-0079-9 Download citation * Received: 03
November 2017 * Revised: 12 February 2018 * Accepted: 10 March 2018 * Published: 01 May 2018 * Issue Date: December 2018 * DOI: https://doi.org/10.1038/s41437-018-0079-9 SHARE THIS ARTICLE
Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided
by the Springer Nature SharedIt content-sharing initiative
Trending News
Tragic tale of malegaon’s supermanBY: GARIMA SHUKLA | Updated Date: Fri, 12 Aug 2011 12:55:00 (IST) इतने साल से हमने सुना कि कई सुपरमेन की जिंदगी का ट्रेज...
Raised latest news in hindi, photos, videos on raised inextlive jagranMeerut News : कूड़ा निपटाने का फार्मूला, कहीं ढेर लगा देते हैैं, कहीं जला देते हैैं local5 months ago अब्दुल्लापुर रोड क...
The uk supreme court's brexit judgment: implications and unintended consequencesThe UK Supreme Court decision in the Brexit litigation on Tuesday is the most important judgment that the Court has deli...
Udder madness: can you see what this fashion brand have removed from tSmooth, blank skin replaces the area where normally women would have nipples. The baffling images have attracted the at...
Manifesto check: conservatives hold the course with schools planThe Conservative Party manifesto makes the following commitments in the area of school-age education: > A good primar...
Latests News
Genomic and geographic footprints of differential introgression between two divergent fish species (solea spp. )ABSTRACT Investigating gene flow between closely related species and its variation across the genome is important to und...
The marketwatch 50 - marketwatchHOW WE CAME UP WITH THE LIST: We asked our readers to submit nominations and the Marketwatch newsroom collected and revi...
Lowe’s scores home run as quarterly sales hold above $20-billionSave for later Lowe’s Cos. Inc. LOW-N on Wednesday backed its expectations for a sales decline in 2021, even after repor...
How to Fight Back Against Age DiscriminationAARP Facebook Twitter LinkedIn Workers who believe their age has cost them — whether it's a job, a promotion, a raise — ...
Mayors push for a new local tax in franceA new ‘residential tax’ should be implemented to help local authorities fund services, a majority of France's mayor...