Pivotal role of bcl11b in the immune, hematopoietic and nervous systems: a review of the bcl11b-associated phenotypes from the genetic perspective
Pivotal role of bcl11b in the immune, hematopoietic and nervous systems: a review of the bcl11b-associated phenotypes from the genetic perspective"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The transcription factor BCL11B plays an essential role in the development of central nervous system and T cell differentiation by regulating the expression of numerous genes
involved in several pathways. Monoallelic defects in the BCL11B gene leading to loss-of-function are associated with a wide spectrum of phenotypes, including neurological disorders with or
without immunological features and susceptibility to hematological malignancies. From the genetic point of view, the landscape of BCL11B mutations reported so far does not fully explain the
genotype-phenotype correlation. In this review, we sought to compile the phenotypic and genotypic variables associated with previously reported mutations in this gene in order to provide a
better understanding of the consequences of deleterious variants. We also highlight the importance of a careful evaluation of the mutation type, its location and the pattern of inheritance
of the variants in order to assign the most accurate pathogenicity and actionability of the genetic findings. SIMILAR CONTENT BEING VIEWED BY OTHERS A MUTANT BCL11B-N440K PROTEIN INTERFERES
WITH BCL11A FUNCTION DURING T LYMPHOCYTE AND NEURONAL DEVELOPMENT Article 01 November 2024 MYELOID NEOPLASMS AND CLONAL HEMATOPOIESIS FROM THE _RUNX1_ PERSPECTIVE Article 30 March 2022 EOMES
IS ESSENTIAL FOR ANTITUMOR ACTIVITY OF CD8+ T CELLS IN CHRONIC LYMPHOCYTIC LEUKEMIA Article Open access 17 March 2021 INTRODUCTION Transcription factors are one of the most potent players
in the regulation of gene expression. The B-cell lymphoma/leukemia 11B gene, BCL11B, is a zinc finger transcription factor first described in chicken in the 2000s by Avram et al. [1], who
reported its pleiotropic functions, involving T-cell maturation, central nervous system development and craniofacial organization. As its paralog BCL11A, BCL11B it is expressed in the
central nervous system, craniofacial tissues and lymphocytes, among other tissues [1, 2]. However, both genes differ in their disease-causing mechanisms. As an example, BCL11A has been found
to be overexpressed in chronic lymphocytic leukemia, immunocytoma and Natural Killer/T-cell lymphoma, suggesting that gain-of-function is the molecular mechanism of pathogenicity [3]. By
contrast, BCL11B haploinsufficiency is associated with T-cell Acute Lymphoblastic Leukemia (T-ALL) and neurological disorders. Curiously, haploinsufficiency of BCL11A is also responsible for
the development of Días-Logan syndrome, a neurological disorder with intellectual disability [4]. BCL11B is placed in the distal region of the chromosome 14, at band 14q32.2. In humans,
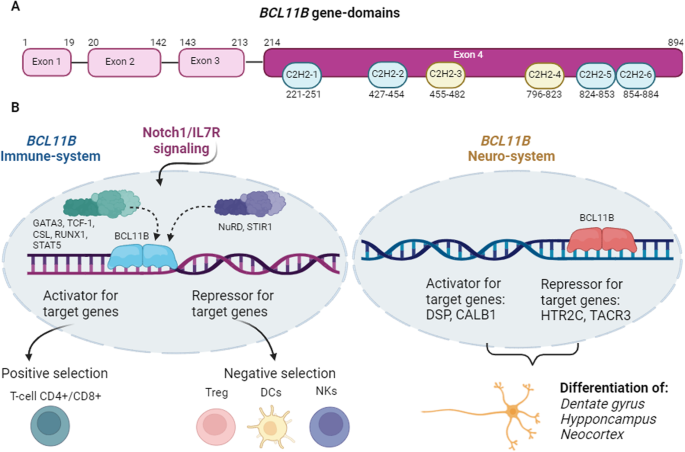
alternative splicing of BCL11B transcripts results in four isoforms, which differ in the presence or absence of exon 3 and the length of the corresponding protein. The reference isoform,
isoform 1 (NM_138576.4; NP_612808.1), contains 894 amino acids and is encoded by four exons, which are organized in six Zinc finger C2H2-type domains [5]. In vitro studies using directed
mutagenesis have elucidated the molecular bases that regulate the function of BCL11B. In this regard, this gene seems to act as a homodimer, in a way that the zinc finger domains are
essential to bind to consensus sites of a variety of genes, such as IL2 and CDKN1A, among others [6, 7]. The third and fourth zinc finger domains are involved in the binding to DNA, whereas
the first, second, fifth and sixth zinc finger domains interact with the proteins MTA1/2, HDAC1/2 [8, 9]. BCL11B possesses activator and repressor properties. Hence, it promotes the
transcription of IL2, cooperating with p300 coactivator, while it also inhibits the transcription of genes such as p57, p21 and SKP2, when it interacts with the nucleosome remodeling and
histone deacetylation complex (NuRD) [7, 10]. This explains why loss of function (LoF) of this integral factor leads to the development of a wide spectrum of diseases. To understand the
genotype-phenotype correlation, we need to understand the function of this gene in specific organs and systems. In this review we have carried out a review covering the different functions
and related phenotypes of BCL11B, describing the landscape of genetic variants and providing keys for the interpretation of the genetic defects in this gene. BCL11B AND THE IMMUNE SYSTEM The
process of BCL11B activation starts when NOTCH1/IL7R signaling promotes the normal activation of BCL11B during thymocyte differentiation with the coordinated action of the transcription
factors CSL, TCF-1 and GATA3, all of which are upregulated by NOTCH-IL7. Once it is activated, the expression of the gene is sustained through the action of RUNX factors [11, 12]. BCL11B is
required at several steps of T-cell precursor differentiation. In the double-negative stage 2 (DN2) T-cell precursors, BCL11B promotes the transition into T-cells α/β T-Cell Receptor (TCR)
through VβDβ rearrangement. It suppresses Natural Killer and dendritic potential in DN2 and DN3 stages and blocks TCR-independent stemness. Consistent with the above, loss of BCL11B function
disrupts the development of T-cell α/β TCR in favor to γ/δ TCR [10]. DN2 T-cells in which BCL11B have been deleted acquire NK cell properties [13]. Knockout BCL11B−/− mice experiment severe
pre-T-cell apoptosis in the thymus, which prevents α/β recombination of VDJ rearrangements at DN stage [14]. Knockout-murine models with complete LoF of BCL11B develop T-cell malignancies,
as a consequence of dysregulated T-cell maturation [15]. In this sense, BCL11B acts as an haploinsufficient tumor suppressor, collaborating in the oncogenic process with other genetic events
[16]. Recently, it has been described a dual role of BCL11B in oncogenesis in such a way that, when overexpressed, it promotes the survival of cancer cells by avoiding cancer cell damage
and cooperating with RAS oncogene in transformation. On the other side, when it is inactivated, its capacity to stimulate several excision repair enzymes is lost, explaining its role as
haploinsufficient tumor suppressor gene [17]. BCL11B exerts a crucial role in the proliferative response of peripheral CD8+ T-cells in response to viruses, intracellular bacteria and tumors.
It regulates CD8 coreceptor gene expression through association with Ei8 and other specific enhancers. Since the coreceptor amplifies TCR signaling, BCL11B defects are associated to reduced
antigen-specific clonal expansion, proliferation and cytolytic function [18]. BCL11B is also linked to autoimmune diseases. This transcription factors induces FOXP3 gene expression in CD4+
cells in response to TGFβ1, by binding to the non-coding sequence. Consequently, defects in BCL11B lead to reduced FOXP3 gene expression and induce Treg generation from conventional CD4+
T-cells. In turn, these Treg cells show reduced expression of IL-10 gene (which is known to play an important role in Treg suppression) and increased levels of proinflammatory cytokines
[19]. BCLL11B AND THE NERVOUS SYSTEM Embryological studies in murine and human models have revealed that BCL11B is expressed in the neocortex, hippocampus, vomeronasal organ and basal
ganglia where is required for the correct development and function of axonal projection of corticospinal motor neurons [20]. It has a critical role in the neurogenesis of the hippocampus,
where it promotes proliferation of progenitor cells and regulates cell differentiation. The absence of BCL11B leads to profound impairment of hippocampus and motoneurons. In adults, BCL11B
is controls the differentiation and survival of mature neurons by up- and down-regulating target genes such as DSP, CALB1, HTR2C and TACR3 [21]. BCL11B IN SKIN DEVELOPMENT AND WOUND HEALING
Murine models have revealed that BCL11B controls the keratinocyte proliferation and late epidermal differentiation events leading to the development of the epidermal permeability barrier.
This effect is mediated by the regulation of specific transcription factors involved in epidermal homeostasis [22]. Increased expression of BCL11B has been described in the basal and
suprabasal layers of the epidermis of patients with atopic dermatitis, contributing to the hyperproliferative state which characterizes this condition [23]. Defects in BCL11B in
keratinocytes lead to altered keratinocyte activation and reduced proliferation through EGFR down regulation. In addition, NFATC1 hyperexpression takes place in BCL111B mutants, suppressing
stem cell activation and delaying wound healing [24]. Consequently, the absence of BCL11B in murine models revealed reduced epithelial thickness in the epidermis and aberrant polarization in
the epithelial cells, which also have consequences for tooth formation and leads to accelerated ossification of skull sutures [22, 25]. The gene structure and molecular pathways involving
immune and neurologic systems regulated by BCL11B are depicted in Fig. 1. BCL11B-ASSOCIATED PHENOTYPES Considering that BCL11B is expressed in multiple organs, tissues and cells, and its
implications in a wide range of cellular processes, it is not surprising that its loss of function results in a complex variability of disorders. However, the correlation between genotype
and phenotype has not been completely understood. In an attempt to classify these phenotypes, several authors have described the following syndromic disorders: immunodeficiency, hematologic
malignancies and neurological disorders. PRIMARY IMMUNODEFICIENCY Severe Combined Immunodeficiency (SCID) associated with monoallelic LoF mutations in BCL11B was first described in 2016 by
Punwani et al. [6]. This syndrome, which begins in the neonatal period, is characterized by T-cell lymphopenia at the expense of CD4+ T cells, impaired T-cell response, and predisposition to
T-ALL. It may be accompanied by dysfunction of B-cells, with defects in the switch from naïve to memory B-cells [6, 26]. Other clinical manifestations include dysmorphic features,
neurodevelopmental abnormalities and a profound immunological dysregulation expressed as allergy, asthma, eczema, eosinophilia and severe atopy [27]. Most of the reported variants were of
germline origin, occurring de novo or being inherited from an affected parent, from which is inferred a dominant inheritance pattern with complete penetrance. HEMATOLOGICAL MALIGNANCIES
Haploinsufficiency of BCL11B contributes to impaired T-cell differentiation and clonal expansion, leading to the development of T-ALL [28]. Truncating and missense variants in this gene have
been reported in 9% of T-ALL patients, with enrichment in mutations located in exon 4 [8]. BCL11B silencing has also been observed as a consequence of structural variants such inv(14) in
T-ALL [29]. Interestingly, chromosomal rearrangements involving BCL11B, have also been found in acute myeloid leukemia (AML) [30]. Variations in BCL11B expression have also been described in
T-ALL, with opposite results. On the one hand it has been demonstrated that overexpression triggers apoptosis resistance [7]. By contrast, low expression was associated with lower survival
in a homogeneously treated population of 169 T-ALL patients [31]. Finally, rare variants in BCL11B have been detected in T-ALL patients accompanying other relevant genetic defects. In a
previous work, we found a missense variant in BCL11B located in a C2H2-type domain in a T-ALL patient accompanied by a SIL-TAL1 rearrangement and deletions in CDKN2A/B [32]. In the same
report, one patient harbored a deleterious variant in BCL11B with an actionable FLT3-internal tandem duplication. In summary, the wide range of potential BCL11B alterations highlights the
central role of this gene in T-ALL leukemogenesis. NEUROLOGICAL DISORDER SCID syndrome caused by BCL11B loss-of-function also presents with severe neurodevelopmental abnormalities and
craniofacial dysmorphism appearing at neonatal age. The most common neurological symptoms include intellectual disability, speech impairment, autistic features and motor delay. Although some
patients may not present obvious immunological abnormalities, intellectual disability is present in 100% of them [33]. The expression of dysmorphic features is variable and may include
myopathic facial appearance, thinning eyebrows, small palpebral fissures, hypertelorism, prominent nose, long philtrum, thin upper lip, ocular and dental abnormalities. Other rare symptoms
reported are dysgenesis of corpus callosum, ventriculomegaly, spasticity of lower limbs and schizophrenia [34, 35]. CRANIOSYNOSTOSIS BCL11B is expressed in cranial osteogenic and sutural
mesenchyme and plays an essential role in the regulation of suture patency. In mice, it has been demonstrated that germline deletion of BCL11B leads to craniofacial synostosis, a disorder
defined by the premature fusion of cranial sutures [36]. In humans, both de novo and inherited mutations result in complex syndromes combining craniosynostosis with immunodeficiency [6],
dysmorphic features, or neurodevelopmental delay [37,38,39]. The mutations in these patients act by disrupting the binding of the protein to its target DNA, which results in
haploinsufficiency [38, 39] or redirecting the protein binding to novel sites [6]. REVIEW OF LITERATURE FROM GENETIC PERSPECTIVE While previous works have described series of patients with
predominant neurological or neoplastic manifestations [16, 26], in our work we have reviewed the whole range of manifestations that may arise as a consequence of genetic defects in BCL11B.
Likewise, we have tried to show that a close relationship exists between the type of mutations, the pattern of inheritance and the phenotype. Finally, we have extended the review of the role
of BCL11B beyond the immune system [2], offering an overview of its role in the development of skin and nervous system. We systematically analyzed PubMed, Medline and EMBASE for all
publications describing BCL11B mutations using the keywords and search terms “BCL11B”; “genetic mutation”/”variant”. Until August 2022, 143 references were retrieved. Publications were
included if they met these criteria: 1) case reports, case series, conferences abstracts and retrospective studies; 2) description of immunodeficiency or neurological disorder or
craniosynostosis or leukemia; 3) The reported mutations were considered by the authors as disease-causing or potentially disease-causing. Table 1 shows the publications selected, together
with the mutations described in each one and the clinical phenotype. The genotypic and phenotypic information was collected and organized according to the following categories: demographics
(sex, age of onset, age at study), clinical data (clinical diagnosis and phenotypes), variant significance (relevant variants, inheritance, allele frequency, in silico prediction, functional
effect and classification of the pathogenicity). All this information is extensively detailed in Supplementary Table 2. Clinical data were grouped into one of these clinical entities:
intellectual developmental disorder with dysmorphic facies, craniosynostosis, T-cell abnormalities and T-ALL (Table 2). This information was completed with the in-silico predictions from six
different sources and the classification of the pathogenicity according to American College of Molecular Genetics criteria (ACMG) [40]. The computational tools allowed us to support a
predicted deleterious/tolerated effect for missense mutations based on alignment of protein sequence, conservation, probability of mutations and models of neural network. The following
bioinformatic predictors were used: Sorting Intolerant from Tolerant (SIFT) [41]; Polymorphism Phenotyping v2 (PolyPhen-2) [42], MutationTaster [43], Combined Annotation Dependent Depletion
(CADD) [44], DANN score (based on a deep neural network) [45] and Functional Analysis through Hidden Markov Models (FATHMM) [46]. Each tool provides a score that indicates the impact or
pathogenicity of the variants on the function of a human protein. The score ranges of the different tools are shown in Supplementary Table 2. In order to classify somatic mutations, the
guidelines of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC) [47] were used. Finally, variants identified in
patients diagnosed with T-ALL in which information regarding their somatic status was missing were searched in the COSMIC database [48]. The variants were annotated based on isoform 1
(NM_138576.4; NP_612808.1). REVIEW OF VARIANTS IN BCL11B ACCORDING TO MUTATION-TYPE (TRUNCATING VERSUS MISSENSE) At least 54 different disease-associated mutations have been reported in the
BCL11B gene. Most of the reported mutations are located in exon 4, with a predominance of nonsynonymous (34/45) over frameshift (14/45) and splicing (1/45) or nonsense (2/45) variants. Seven
mutations have been described, in exon 2, four of them frameshift, one deleterious and one nonsynonymous. Only one mutation has been described in exon one and other in exon three. Details
regarding these mutations are shown in Table 1 and Fig. 1. The mutations were identified in 62 patients, of whom 24 harbored truncating mutations and 38 missense variants. Since it has been
demonstrated that monoallelic LoF variants are responsible for a BCL11B-related phenotype, all truncating mutations found in the sequence of BCL11B have been classified as pathogenic or
likely pathogenic. Only four functional studies have proved loss of function of missense mutations. In the remaining, missense variants, either functional studies or robust logarithm of the
odds would be required to classify them as pathogenic. TRUNCATING VARIANTS EXON 2 A frameshift variant located in exon 2, p.Cys81Leufs*76, was predicted to suffer nonsense-mediated mRNA
decay (NMD) and non-stop mRNA decay (NMD), resulting in haploinsufficiency of the protein [26]. The variant p.Ala891Profs*106, placed in the last coding residue, was predicted to extend the
protein translation beyond the last coding residue, suffering non-stop mRNA decay and haploinsufficiency of the protein, too. In the same study, seven truncating variants located in exon 4
(p.Tyr455*, p.Glu499*, p.Thr502Hisfs*15, p.Arg518Alafs*45, p.Asp534Thrfs*29, p.Gly649Alafs*67 and p.Gly820Alafs*27) were assumed to escape this mechanism of mRNA degradation. These changes
are highly likely to result in proteins lacking the last Zinc-finger domains. However, only the p.Gly820Alafs*27 variant was tested to assess the stability of the allele carrying the
mutation. Immunohistological staining showed that no stable cDNA was expressed in the mutant compared to the wild-type allele. Real-time PCR was not done in the other variants to confirm the
predicted haploinsufficiency effect. The only splicing variant presented in this review, c.427+1G>A in exon 2 (observed in three patients from same family), produced the exon skipping
during the splicing process, leading to the introduction of a premature stop codon at p.Val48Glyfs∗14, before C2H2-1 finger domain. The aberrant transcript caused by this variant was
supposed to be degraded by NMD, but western blotting and immunofluorescence assays revealed normal expression and no significant changes in cell morphology or localization [49], suggesting
the existence of post-transcriptional regulatory mechanisms affecting protein function as the disease-causing mechanism of the mutation. EXON 4 According to the algorithm of the
computational tool AlphaFold, the frameshift mutation p.Gly783Alafs*24, placed the stop codon in the C2H2-4 domain in exon 4. It was also predicted to escape NMD. This truncated protein
would lack the last three C2H2 zinc-finger domains and hence affect the protein´s function in binding to its target DNA. However, the authors recommend further experiments to elucidate the
true mechanisms of action of the variant [38]. Regarding the insertion c.2461_2462ins (p.Glu821Glyfs∗28), located in the exon 4, the results observed in the qPCR assay showed no difference
in the level of mRNA between mutant and wild-type transfected cells. Therefore, the scape of this variant form NMD was confirmed. This variant loses the last two C-terminal DNA binding
Zinc-finger domains, probably resulting in a truncated protein with a pathogenic mechanism of haploinsufficiency [50]. The variants p.Ser398Glnfs∗117, p.Gly630Thrfs*91 and p.Thr730Thrfs*151
were described in de novo case reports [33, 50, 51]. According to the position of the premature stop codon, these three variants were expected to escape nonsense‐mediated mRNA decay. This
would lead to a truncating protein, conserving the first zinc finger domain in the variant p.Ser398Glnfs*117 and the first, second and third finger domains in the case of the variants
p.Gly630Thrfs*91 and p.Thr730Thrfs*151. Six truncating variants (p.Leu30fs*, p.Asp33fs*, p.Leu36_Glu37fs*, p.Thr120fs* located in exon 2 and p.Tyr384*, p.Thr450fs* located in exon 4) were
identified only in patients with T-ALL; Surprisingly, in none of them functional studies were performed and neither had confirmed zygosity [52, 53]. Nevertheless, p.Asp33fs* and
p.Leu36_Glu37fs* were reported in COSMIC database as somatic mutations in specimens with T-ALL. Although the molecular mechanism of pathogenicity was not studied, the variants p.Thr120fs*,
p.Tyr384*, p.Thr450fs* would scape NMD, while the rest will directly suffer NMD. MISSENSE VARIANTS Thirty-eight patients harbored 33 unique non-synonymous mutations located throughout the
four exons. The most frequent diagnosis was T-ALL in 28 patients, whereas neurological disorders were present in 10 of the 38 patients. Among the latter, 4 showed concomitant immunological
disorders. Germline origin was confirmed in patients with neurological disorders. Amino acid substitutions secondary to missense mutation were the most frequent defect in T-ALL. Regarding
these missense variants, all but 3 SNVs had a maximum allele frequency of <0.005%, suggesting that this gene is very conserved, in line with the function of transcription factors which
maintain high fidelity in their aminoacidic sequence especially in DNA-binding motifs. EXON 1 The p.Arg3Ser variant was the only reported mutation located within the first exon of BCL11B
(Table 1 and Fig. 1). This missense variant occurred de novo in a patient with seizures, dysmorphic features and craniosynostosis. No immunological alterations were observed. The variant was
absent in the control population and 5 out of 6 bioinformatics tools predicted a deleterious effect. The functional study of p.Arg3Ser in HEK293T-cells showed normal expression and
localization of BCL11B mutants, but inability of the protein to interact both with the NuRD and PRC2 complexes. The substitution of Arg3 changed the charge of the “RRKQxxP” motif, generating
a novel potential site for post-translational modification suggesting alternative positions at nearby residues [35]. Specifically, this variant causes a change from an amino acid with a
basic side chain (arginine) to an amino acid with a neutral polar side chain (serine). Finally, a mouse model for BCL11B p.Arg3Ser mutants showed coronal suture craniosynostosis in both
heterozygotes and homozygotes, with a less severe and more variable phenotype in heterozygotes. EXON 2 The p.His126Tyr variant, located in exon 2, was reported in a patient with ETP-ALL as a
rare finding together with a pathogenic mutation in the FLT3 gene. This variant presented an alternative allele frequency of 50%, suggesting germline origin. In fact, this change had 9
heterozygotes in the general population. However, since it has not been confirmed in non-hematological tissues, the germline origin should not be definitely considered [32]. EXON 3 A
non-described variant, p.Gln238His (p.Gln167His) located in exon 3, was detected in an adult patient with T-ALL. No functional studies were performed [53]. EXON 4 Germline mutations
p.Pro422Leu, p.Gly582Ser, p.Gly667Glu, and p.Pro673Arg were found in patients with craniosynostosis and congenital diaphragmatic hernia. In all cases parents were unaffected, suggesting
incomplete penetrance [54]. They present in a very low frequency in general population and are located in a hotspot region for pathogenic mutations. The in-silico predictor prognosed a
likely tolerate effect. Therefore, they should be considered variants of unknown significance. Further functional studies would be required to clarify the pathogenicity of the mutations.
Numerous missense mutations situated in exon 4 (p.Cys432Arg, p.Arg447Cys, p.Glu452Lys, p.Pro454Thr, p.Tyr455Asn, p.Tyr455His, p.Tyr455Cys, p.Ser465Leu, p.Gln466His, p.Lys469Thr, p.Arg472Cys,
p.Arg472His, p.Lys475Glu, p.Gly581Asn, p.Gly596Ser, p.Ala834Thr, p.Gln835His, p.Gly847Arg and p.Gln848Arg) have been identified in pediatric or young patients diagnosed with T-ALL, with a
deleterious effect according to computational results and a very low allele frequency or even absence in the general population [16, 32, 52]. All of them were concentrated in the C2H2-2/3
and 4 domains, which are considered a hot-spot for pathogenic mutations in BCL11B. Using high-resolution crystal structure of the zinc finger domains and based on models with structural
homology of canonical DNA binding of BCL11B, Gutierrez et al. [16] evaluated four missense variants (p.His445Tyr, p.Arg447His, p.Arg472His, p.His479Tyr) located withing this hot-spot in exon
4. The results showed that the affected residues disrupted DNA binding by destabilization of the conserved amino acids. Although a functional study of p.Thr450Met was not performed, it was
recurrently found in patients with T-ALL with poor prognosis [32, 52, 55]. Considering its low prevalence in the general population and its zygosity compatible with heterozygous state
(allele frequency close to 50%), it might be argued that this change is a rare germline variant with a high potential of having a deleterious effect on BCL11B. The variants p.Asn807Lys [26]
and p.Cys826Tyr [21] require special attention because of their publication in two unrelated patients as de novo. Both are located in exon 4 and affect domains C2H2-4 and C2H2-5,
respectively. The p.Asn807Lys variant was predicted to affect interaction between specific residues of the C2H2-4 domain and DNA. The p.Cys826Tyr variant was evaluated by structural
modeling, using the crystal structure of the related transcription factor BCL11A. This model revealed that the change could perturb the coordination of zinc ions, thereby affecting protein
interaction [27]. A functional study in a zebrafish model showed that mutant T-cells were unable to differentiate to terminal states. Patients harboring these mutations also showed
neurological disorders with dysmorphic features and T-cell disturbances such as low Treg counts and hyper IgE syndrome. No mutations have been described in exons first and sixth Zinc finger
domains. Figure 2 shows the mutations located along the BCL11B gene. GENOTYPE-PHENOTYPE CORRELATION The probability of being Loss-of-function-Intolerant (pLI) of BCL11B is 0.99, suggesting
that this gene would not tolerate the presence of variants with LoF effects such as nonsense, frameshift and splicing [56,57,58]. In fact, LoF mutations in BCL11B cause two major clinical
pictures that do not overlap: T-ALL and neurological disorders. Recurrent infections and immunological disturbances may be present in the last group. From the review it might be suggested
that a relationship between the phenotype and the mutation-type could be stablished; 28 out of 34 T-ALL patients harbored missense mutations compared to 10 out of 28 patients with
neurological disorders who harbored missense mutations. In other words, missense mutations are responsible for the majority of T-ALL whereas truncating mutations predominate in patients with
neurological disorders. The distribution of the type of mutations according to the clinical phenotype is shown in Table 2. In addition, we compared the correlation between genetic variables
such as mutation type and inheritance status and phenotype by using the Pearson’s correlation coefficient. The results of this comparison are shown in Fig. 3 and shows the association
between germline truncating mutations and neurological disorders and somatic missense mutations and T-ALL, respectively. More detailed information regarding the pattern of inheritance is
shown in Supplementary Table 2. DISCUSSION In this review we have gathered information about mutations in BCL11B published to date. We have focused on potentially disease-causing mutations
and extensively reviewed their molecular structure and their functional and clinical consequences. Some of these mutations, particularly truncating mutations, had been classified as
mutations of uncertain significance according to ACMG criteria. However, after applying bioinformatic predictors, we suggest that most of them could be reclassified as pathogenic. Our review
was also aimed to clarify the contexts that need to be considered when interpreting the consequences of the reported BCL11B variants. Specifically, the type of the mutations (truncating or
missense), the location in the domains of the protein and the level of expression in the affected tissue and the pattern of inheritance (germline or somatic) must be considered. In general,
truncating mutations are associated to neurodevelopmental disorders with dysmorphic features and immunologic disturbances. Rare missense variants causing BCL11B haploinsufficiency are also
responsible for some cases of neurological disorders. It is interesting that no missense mutations were reported in patients with neurological disorders in exons 2 and 3, and only two
missense variants placed in these two exons were found in patients with craniosynostosis and T-ALL, which suggests that this region is not a hot-spot. On the other hand, missense mutations
and deletions are associated with T-ALL. It may be hypothesized that such skewed distribution in the type of mutations related to the phenotype may be explained by the level of expression of
BCL11B in the affected tissues and that epigenetic regulation of the expression is responsible for the differences. In line with this argument, hypomorphic missense mutations in BCL11B
express variable features, different to the ones observed in truncating mutations. Regarding the pattern of inheritance, it is striking that 100% of the disease-causing variants in
BCL11B-neurological disorders were of germline origin, either de novo or inherited from an affected parent. In contrast, nearly all the missense variants detected in T-ALL patients were
somatic or of unknown origin. An exception to this rule is a recently reported mutation, p.Thr450Met, considered of probable germline origin. This heritability of the disease has obvious
clinical implications when selecting donor from siblings in case of performing hematopoietic stem cell transplant. The lack of information regarding zygosity from missense variants in
patients with T-ALL constrains the possibility of estimate the genotype-phenotype correlation as well as the functional consequences of the variants. Finally, there were no reported patients
with missense germline mutations with immunological abnormalities as the sole manifestation of the disease, i.e. without extra-immune manifestations. Something similar happened in other
restricted phenotypes associated with LoF mutations in BCL11B, such is craniosynostosis without other symptoms or milder features. Mutations causing immunological abnormalities such as low
peripheral blood T lymphocyte counts, dysfunctional type 2 innate lymphoid cells and autoimmunity, do not share a common pattern, making it difficult to predict the effect of such mutations.
The plasticity of the immune system could explain the modulation in the expressiveness of the BCL11B defects. In fact, the variability of the expression is comparable to that observed in
other diseases caused by defects in transcription factors with a wide range of target genes, such as IKZF1 or GATA2 [59]. Although we have provided an extensive analysis about the
genotype-phenotype correlation, there are several shortcomings that limit the generalizability of the findings. First, the reduced number of patients analyzed; second, the heterogeneity of
their diseases and finally, the lack of functional studies, especially regarding missense mutations. Therefore, it is desirable that future reports include functional studies proving the
pathogenicity of the variants. CONCLUSIONS In summary, the relevance of the BCL11B gene is reflected in the wide range of disorders that arise as a consequence of its loss of function. In
order to stablish the pathogenicity of the genetic variants of BCL11B and the heterogeneity of the clinical manifestations, several aspects must be considered, including the type of
mutation, the position of premature stop codon, the inheritance status and the function of specific domains. The precise interpretation of the genetic defect in BCL11B may guide the
development of targeted treatments, especially, in the context of genome editing. In this sense, attempts of restoring BCL11B function with antisense oligonucleotides in animal models of
SCID have been made and pave the way for genome editing [6].Considering that BCL11B haploinsufficiency has leukemogenic effects, further studies should also address the regulation of BCL11B
expression at the molecular level. Finally, functional data could elucidate the consequences of nonsynonymous variants in the BCL11B gene. REFERENCES * Avram D, Fields A, Pretty On Top K,
Nevrivy DJ, Ishmael JE, Leid M. Isolation of a novel family of C(2)H(2) zinc finger proteins implicated in transcriptional repression mediated by chicken ovalbumin upstream promoter
transcription factor (COUP-TF) orphan nuclear receptors. J Biol Chem. 2000;275:10315–22. Article CAS PubMed Google Scholar * Avram D, Califano D. The multifaceted roles of Bcl11b in
thymic and peripheral T cells: impact on immune diseases. J Immunol. 2014;193:2059–65. Article CAS PubMed Google Scholar * Satterwhite E, Sonoki T, Willis TG, Harder L, Nowak R, Arriola
EL, et al. The BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood. 2001;98:3413–20. Article CAS PubMed Google Scholar * Dias C, Estruch SB, Graham SA, McRae J,
Sawiak SJ, Hurst JA, et al. BCL11A Haploinsufficiency Causes an Intellectual Disability Syndrome and Dysregulates Transcription. Am J Hum Genet. 2016;99:253–74. Article CAS PubMed PubMed
Central Google Scholar * Lennon MJ, Jones SP, Lovelace MD, Guillemin GJ, Brew BJ. Bcl11b: A New Piece to the Complex Puzzle of Amyotrophic Lateral Sclerosis Neuropathogenesis? Neurotox
Res. 2016;29:201–7. Article CAS PubMed Google Scholar * Punwani D, Zhang Y, Yu J, Cowan MJ, Rana S, Kwan A, et al. Multisystem Anomalies in Severe Combined Immunodeficiency with Mutant
BCL11B. N Engl J Med. 2016;375:2165–76. Article CAS PubMed PubMed Central Google Scholar * Grabarczyk P, Nähse V, Delin M, Przybylski G, Depke M, Hildebrandt P, et al. Increased
expression of bcl11b leads to chemoresistance accompanied by G1 accumulation. PLoS One. 2010;5:e12532. Article PubMed PubMed Central Google Scholar * Huang X, Du X, Li Y. The role of
BCL11B in hematological malignancy. Exp Hematol Oncol. 2012;1:22. Article CAS PubMed PubMed Central Google Scholar * Kominami R. Role of the transcription factor Bcl11b in development
and lymphomagenesis. Proc Jpn Acad Ser B Phys Biol Sci. 2012;88:72–87. Article CAS PubMed PubMed Central Google Scholar * Cismasiu VB, Adamo K, Gecewicz J, Duque J, Lin Q, Avram D.
BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene. 2005;24:6753–64. Article CAS PubMed Google Scholar * Kueh HY, Yui MA, Ng KK,
Pease SS, Zhang JA, Damle SS, et al. Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat Immunol. 2016;17:956–65. Article CAS PubMed
PubMed Central Google Scholar * Georgopoulos K. Induction of Bcl11b during T cell commitment through a tripartite mechanism. Nat Immunol. 2016;17:903–4. Article CAS PubMed Google
Scholar * Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC, et al. Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science. 2010;329:85–9. Article CAS PubMed
PubMed Central Google Scholar * Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, et al. Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat Immunol.
2003;4:533–9. Article CAS PubMed Google Scholar * Ehrlich LA, Yang-Iott K, Bassing CH. Tcrδ translocations that delete the Bcl11b haploinsufficient tumor suppressor gene promote
atm-deficient T cell acute lymphoblastic leukemia. Cell Cycle. 2014;13:3076–82. Article CAS PubMed PubMed Central Google Scholar * Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC,
et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood. 2011;118:4169–73. Article CAS PubMed PubMed Central
Google Scholar * Vickridge E, Faraco CCF, Lo F, Rahimian H, Liu ZY, Tehrani PS, et al. The function of BCL11B in base excision repair contributes to its dual role as an oncogene and a
haplo-insufficient tumor suppressor gene. Nucleic Acids Res. 2023:gkad1037. https://doi.org/10.1093/nar/gkad1037. * Zhang S, Rozell M, Verma RK, Albu DI, Califano D, VanValkenburgh J, et al.
Antigen-specific clonal expansion and cytolytic effector function of CD8+ T lymphocytes depend on the transcription factor Bcl11b. J Exp Med. 2010;207:1687–99. Article CAS PubMed PubMed
Central Google Scholar * Vanvalkenburgh J, Albu DI, Bapanpally C, Casanova S, Califano D, Jones DM, et al. Critical role of Bcl11b in suppressor function of T regulatory cells and
prevention of inflammatory bowel disease. J Exp Med. 2011;208:2069–81. Article CAS PubMed PubMed Central Google Scholar * Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis
JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–21. Article CAS PubMed Google Scholar * Simon R, Baumann L, Fischer
J, Seigfried FA, De Bruyckere E, Liu P, et al. Structure-function integrity of the adult hippocampus depends on the transcription factor Bcl11b/Ctip2. Genes Brain Behav. 2016;15:405–19.
Article CAS PubMed Google Scholar * Golonzhka O, Liang X, Messaddeq N, Bornert JM, Campbell AL, Metzger D, et al. Dual role of COUP-TF-interacting protein 2 in epidermal homeostasis and
permeability barrier formation. J Invest Dermatol. 2009;129:1459–70. Article CAS PubMed Google Scholar * Ganguli-Indra G, Liang X, Hyter S, Leid M, Hanifin J, Indra AK. Expression of
COUP-TF-interacting protein 2 (CTIP2) in human atopic dermatitis and allergic contact dermatitis skin. Exp Dermatol. 2009;18:994–6. Article CAS PubMed PubMed Central Google Scholar *
Liang X, Bhattacharya S, Bajaj G, Guha G, Wang Z, Jang HS, et al. Delayed cutaneous wound healing and aberrant expression of hair follicle stem cell markers in mice selectively lacking Ctip2
in epidermis. PLoS One. 2012;7:e29999. Article CAS PubMed PubMed Central Google Scholar * Daher MT, Bausero P, Agbulut O, Li Z, Parlakian A. Bcl11b/Ctip2 in Skin, Tooth, and
Craniofacial System. Front Cell Dev Biol. 2020;8:581674. Article PubMed PubMed Central Google Scholar * Lessel D, Gehbauer C, Bramswig NC, Schluth-Bolard C, Venkataramanappa S, van
Gassen KLI, et al. BCL11B mutations in patients affected by a neurodevelopmental disorder with reduced type 2 innate lymphoid cells. Brain. 2018;141:2299–311. Article PubMed PubMed Central
Google Scholar * Lu HY, Sertori R, Contreras AV, Hamer M, Messing M, Del Bel KL, et al. A Novel Germline Heterozygous BCL11B Variant Causing Severe Atopic Disease and Immune
Dysregulation. Front Immunol. 2021;12:788278. Article CAS PubMed PubMed Central Google Scholar * Go R, Hirose S, Morita S, Yamamoto T, Katsuragi Y, Mishima Y, et al. Bcl11b
heterozygosity promotes clonal expansion and differentiation arrest of thymocytes in gamma-irradiated mice. Cancer Sci. 2010;101:1347–53. Article CAS PubMed PubMed Central Google Scholar
* Przybylski GK, Dik WA, Wanzeck J, Grabarczyk P, Majunke S, Martin-Subero JI, et al. Disruption of the BCL11B gene through inv(14)(q11.2q32.31) results in the expression of BCL11B-TRDC
fusion transcripts and is associated with the absence of wild-type BCL11B transcripts in T-ALL. Leukemia. 2005;19:201–8. Article CAS PubMed Google Scholar * Bezrookove V, van
Zelderen-Bhola SL, Brink A, Szuhai K, Raap AK, Barge R, et al. A novel t(6;14)(q25-q27;q32) in acute myelocytic leukemia involves the BCL11B gene. Cancer Genet Cytogenet. 2004;149:72–6.
Article CAS PubMed Google Scholar * Bartram I, Gökbuget N, Schlee C, Heesch S, Fransecky L, Schwartz S, et al. Low expression of T-cell transcription factor BCL11b predicts inferior
survival in adult standard risk T-cell acute lymphoblastic leukemia patients. J Hematol Oncol. 2014;7:51. Article PubMed PubMed Central Google Scholar * García-Aznar JM, Alonso S,
Iglesias DU, de Ugarriza PL, López CÁ, Balbín M, et al. Mapping the genetic features of T-ALL cases through simplified NGS approach. Clin Immunol. 2022;245:109151. Article PubMed Google
Scholar * Qiao F, Wang C, Luo C, Wang Y, Shao B, Tan J, et al. A De Novo heterozygous frameshift mutation identified in BCL11B causes neurodevelopmental disorder by whole exome sequencing.
Mol Genet Genom Med. 2019;7:e897. Article Google Scholar * Prasad M, Balci TB, Prasad C, Andrews JD, Lee R, Jurkiewicz MT, et al. BCL11B-related disorder in two canadian children:
Expanding the clinical phenotype. Eur J Med Genet. 2020;63:104007. Article CAS PubMed Google Scholar * Fahey L, Donohoe G, Broin PÓ, Morris DW. Genes regulated by BCL11B during T-cell
development are enriched for de novo mutations found in schizophrenia patients. Am J Med Genet B Neuropsychiatr Genet 2020;183:370–9. Article CAS PubMed Google Scholar * Kyrylkova K,
Iwaniec UT, Philbrick KA, Leid M. BCL11B regulates sutural patency in the mouse craniofacial skeleton. Dev Biol. 2016;415:251–60. Article CAS PubMed Google Scholar * Goos JAC, Vogel WK,
Mlcochova H, Millard CJ, Esfandiari E, Selman WH, et al. A de novo substitution in BCL11B leads to loss of interaction with transcriptional complexes and craniosynostosis. Hum Mol Genet.
2019;28:2501–13. Article CAS PubMed PubMed Central Google Scholar * Zhao X, Wu B, Chen H, Zhang P, Qian Y, Peng X, et al. Case report: A novel truncating variant of BCL11B associated
with rare feature of craniosynostosis and global developmental delay. Front Pediatr. 2022;10:982361. Article PubMed PubMed Central Google Scholar * Eto K, Machida O, Yanagishita T,
Shimojima Yamamoto K, Chiba K, Aihara Y, et al. Novel BCL11B truncation variant in a patient with developmental delay, distinctive features, and early craniosynostosis. Hum Genome Var.
2022;9:43. Article CAS PubMed PubMed Central Google Scholar * Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of
sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. Article
PubMed PubMed Central Google Scholar * Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids
Res. 2012;40:W452–7. Article CAS PubMed PubMed Central Google Scholar * Adzhubei I, Jordan DM, Sunyaev SR Predicting functional effect of human missense mutations using PolyPhen-2. Curr
Protoc Hum Genet. 2013;Chapter 7:Unit7.20. https://doi.org/10.1002/0471142905.hg0720s76. * Steinhaus R, Proft S, Schuelke M, Cooper DN, Schwarz JM, Seelow D. MutationTaster2021. Nucleic
Acids Res. 2021;49:W446–51. Article CAS PubMed PubMed Central Google Scholar * Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the
relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. Article CAS PubMed PubMed Central Google Scholar * Quang D, Chen Y, Xie X. DANN: a deep learning approach for
annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31:761–3. Article CAS PubMed Google Scholar * Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et
al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. Article CAS PubMed Google Scholar
* Horak P, Griffith M, Danos AM, Pitel BA, Madhavan S, Liu X, et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations
of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet Med. 2022;24:986–98. Article CAS PubMed PubMed
Central Google Scholar * Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–7. Article
CAS PubMed Google Scholar * Che F, Tie X, Lei H, Zhang X, Duan M, Zhang L, et al. Identification of two novel variants of the BCL11B gene in two Chinese pedigrees associated with
neurodevelopmental disorders. Front Mol Neurosci. 2022;15:927357. Article CAS PubMed PubMed Central Google Scholar * Yan S, Wei YS, Yang QY, Yang L, Zeng T, Tang XM, et al. [A case
report of BCL11B mutation induced neurodevelopmental disorder and literature review]. Zhonghua Er Ke Za Zhi 2020;58:223–7. CAS PubMed Google Scholar * Yang S, Kang Q, Hou Y, Wang L, Li L,
Liu S, et al. Mutant BCL11B in a Patient With a Neurodevelopmental Disorder and T-Cell Abnormalities. Front Pediatr. 2020;8:544894. Article PubMed PubMed Central Google Scholar * Liu Y,
Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–8. Article
CAS PubMed PubMed Central Google Scholar * Neumann M, Vosberg S, Schlee C, Heesch S, Schwartz S, Gökbuget N, et al. Mutational spectrum of adult T-ALL. Oncotarget. 2015;6:2754–66.
Article PubMed Google Scholar * Gaillard L, Goverde A, van den Bosch QCC, Jehee FS, Brosens E, Veenma D, et al. Case Report and Review of the Literature: Congenital Diaphragmatic Hernia
and Craniosynostosis, a Coincidence or Common Cause? Front Pediatr. 2021;9:772800. Article PubMed PubMed Central Google Scholar * Seki M, Kimura S, Isobe T, Yoshida K, Ueno H,
Nakajima-Takagi Y, et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet. 2017;49:1274–81. Article CAS PubMed Google Scholar * Lek M,
Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. Article CAS PubMed PubMed
Central Google Scholar * Ziegler A, Colin E, Goudenège D, Bonneau D. A snapshot of some pLI score pitfalls. Hum Mutat. 2019;40:839–41. PubMed Google Scholar * Kuehn HS, Nunes-Santos CJ,
Rosenzweig SD. Germline IKZF1 mutations and their impact on immunity: IKAROS-associated diseases and pathophysiology. Expert Rev Clin Immunol. 2021;17:407–16.
https://doi.org/10.1080/1744666X.2021.1901582. Article CAS PubMed PubMed Central Google Scholar * Calvo KR, Hickstein DD. The spectrum of GATA2 deficiency syndrome. Blood.
2023;141:1524–32. Article CAS PubMed Google Scholar Download references AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Healthincode, A Coruña, Spain José María García-Aznar * Universitary
Institute of Oncology of Principado de Asturias (IUOPA), Oviedo, Spain José María García-Aznar, Sara Alonso Alvarez & Teresa Bernal del Castillo * Health Research Institute of
Principado de Asturias, Oviedo, Spain José María García-Aznar, Sara Alonso Alvarez & Teresa Bernal del Castillo * Hematology Department, Hospital Universitario Clínico de Asturias,
Oviedo, Spain Sara Alonso Alvarez & Teresa Bernal del Castillo Authors * José María García-Aznar View author publications You can also search for this author inPubMed Google Scholar *
Sara Alonso Alvarez View author publications You can also search for this author inPubMed Google Scholar * Teresa Bernal del Castillo View author publications You can also search for this
author inPubMed Google Scholar CONTRIBUTIONS JGA contributed to the concept, writing, and editing the manuscript. SAA contributed to the supervision of the manuscript. TGC contributed to the
writing, supervision and editing the manuscript. CORRESPONDING AUTHOR Correspondence to José María García-Aznar. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing
interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY TABLE 1. SUPPLEMENTARY TABLE 2 SUPPLEMENTARY LIST_OF_ABBREVIATIONS RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution
4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and
the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s
Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not
permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE García-Aznar, J.M., Alonso Alvarez, S. & Bernal del Castillo, T. Pivotal role
of BCL11B in the immune, hematopoietic and nervous systems: a review of the BCL11B-associated phenotypes from the genetic perspective. _Genes Immun_ 25, 232–241 (2024).
https://doi.org/10.1038/s41435-024-00263-w Download citation * Received: 22 August 2023 * Revised: 19 February 2024 * Accepted: 22 February 2024 * Published: 12 March 2024 * Issue Date: June
2024 * DOI: https://doi.org/10.1038/s41435-024-00263-w SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable
link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Drivers urged to pay car tax ahead of major ved changes next monthThe standard rate will increase by £10 for most cars which were first registered on or after April 1, 2017. For cars reg...
First baby due in new ivf technique | nursing timesThe first baby to be conceived using a breakthrough IVF technique will be born in Scotland next month, a clinic has said...
Wimbledon To Lose Familiar Face As Veteran BBC Presenter Sue Barker Calls Time After 30 YearsSue Barker at WimbledonBBC This year’s Wimbledon tennis tournament will see the end of an era. No, Roger Federer has not...
Elevation and fog-cloud similarity in tibeto-burman languagesABSTRACT Lexically, 52.99% of the Tibeto-Burman languages, the non-Sinitic branches of the Sino-Tibetan language family,...
Children's hospice launches trailblazing transition support role | nursing timesA charity that provides hospice care to seriously ill children in the North West of England is blazing a trail in the se...
Latests News
Pivotal role of bcl11b in the immune, hematopoietic and nervous systems: a review of the bcl11b-associated phenotypes from the genetic perspectiveABSTRACT The transcription factor BCL11B plays an essential role in the development of central nervous system and T cell...
[Short Reviews] | NatureABSTRACT THIS book has been specially prepared for secondary school pupils taking advanced courses in science and engine...
[Obituaries] | NatureABSTRACT WE regret to announce the following deaths: ARTICLE PDF ENJOYING OUR LATEST CONTENT? LOGIN OR CREATE AN ACCOUNT...
Droplet genealogy | Nature PhysicsThe tendency of a stationary droplet, sitting on the surface of a large body of liquid, to eject a smaller droplet when ...
Pest Infestation of Produce | NatureABSTRACT THE Secretary of the Department of Scientific and Industrial Research announces that H.M. Government has gratef...