Arhgap35 is a novel factor disrupted in human developmental eye phenotypes
Arhgap35 is a novel factor disrupted in human developmental eye phenotypes"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT ARHGAP35 has known roles in cell migration, invasion and division, neuronal morphogenesis, and gene/mRNA regulation; prior studies indicate a role in cancer in humans and in the
developing eyes, neural tissue, and renal structures in mice. We identified damaging variants in _ARHGAP35_ in five individuals from four families affected with anophthalmia, microphthalmia,
coloboma and/or anterior segment dysgenesis disorders, together with variable non-ocular phenotypes in some families including renal, neurological, or cardiac anomalies. Three variants
affected the extreme C-terminus of the protein, with two resulting in a frameshift and C-terminal extension and the other a missense change in the Rho-GAP domain; the fourth (nonsense)
variant affected the middle of the gene and is the only allele predicted to undergo nonsense-mediated decay. This study implicates _ARHGAP35_ in human developmental eye phenotypes.
C-terminal clustering of the identified alleles indicates a possible common mechanism for ocular disease but requires further studies. SIMILAR CONTENT BEING VIEWED BY OTHERS DELETION
UPSTREAM OF _MAB21L2_ HIGHLIGHTS THE IMPORTANCE OF EVOLUTIONARILY CONSERVED NON-CODING SEQUENCES FOR EYE DEVELOPMENT Article Open access 26 October 2024 _GJA8_-ASSOCIATED DEVELOPMENTAL EYE
DISORDERS: A NEW MULTICENTRE STUDY HIGHLIGHTS MUTATIONAL HOTSPOTS AND GENOTYPE-PHENOTYPE CORRELATIONS Article Open access 30 April 2025 LOSS OF GAP JUNCTION DELTA-2 (_GJD2_) GENE ORTHOLOGS
LEADS TO REFRACTIVE ERROR IN ZEBRAFISH Article Open access 03 June 2021 INTRODUCTION Developmental ocular disorders including microphthalmia, anophthalmia, and coloboma (MAC) and anterior
segment dysgenesis (ASD) spectrums represent an important cause of vision loss in childhood. Genetic diagnoses are unable to be established in a significant portion of cases: 70–85% of MAC
and 40–75% of ASD cases remain unexplained after genetic analysis according to recent studies [1,2,3,4]. While some have idiopathic or environmental causes, others are likely caused by
pathogenic variants in genes with a currently unrecognized role in eye development. Establishing a genetic diagnosis is important for clinical management of affected children and provides
other family members with the opportunity to clarify their genetic status/risk. Identification of novel genetic causes of ocular disorders enhances our understanding of the mechanisms of
ocular development, generating additional opportunities for therapeutic intervention in the future. ARHGAP35 (Rho GTPase Activating Protein 35), also known as GRLF1 (Glucocorticoid Receptor
DNA-binding Factor 1) or p190ARhoGAP-A, is a GTPase-activating protein that regulates GTPases within the Rho and Rac families [5, 6]. It has known direct roles in cell migration/invasion and
division as well as neuronal morphogenesis and dendritic spine formation; additional roles include gene/mRNA translation regulation through interaction with TFII-I and eiF3A [5]. Mice
homozygous for a loss-of-function _Arhgap35_ allele display highly penetrant early lethality, structural brain anomalies, cystic glomeruli, and optic cup anomalies including coloboma and
microphthalmia while heterozygous mice are unaffected; a subset of homozygous animals also display neural tube closure defects, particularly exencephaly [7, 8]. Little is known about the
role of germline variants in humans; only one de novo missense variant, c.1801G > T p.(Val601Phe), has been reported in the literature: limited phenotypic description reported a
terminated pregnancy with severe midline birth defects [9]. The rate of de novo variants in _ARHGAP35_ was also noted to be enriched in a large cohort of individuals with developmental
disorders with no details provided [10]. Through exome/genome sequencing and GeneMatcher, we identified a cohort of individuals with developmental ocular disorders and novel damaging
variants in _ARHGAP35_. MATERIALS AND METHODS This human study was approved by Institutional Review Boards at Children’s Wisconsin and Einstein Medical Center Philadelphia. Written informed
consent including research analysis and photo publication if applicable was obtained for every participant. Exome sequencing was undertaken by Psomagen (Rockville, MD) and analyzed with
VarSeq (Golden Helix, Bozeman, MT). In silico analysis of variants of interest included filtering for frequency <0.001 in the general population in gnomAD v2.1.1 [11] and for predicted
effect upon the protein. The effect of missense variants on protein function was further analyzed by two combined analysis tools (CADD phred hg19 and REVEL). Samples were first analyzed for
variants in known MAC and ASD genes as previously described [12, 13]. Trio analysis in negative cases identified _ARHGAP35_ as a candidate in two families and screening for variants in this
gene specifically identified one more case. Sanger sequencing was used to confirm variants and for segregation analysis. An additional case was identified through clinical genome sequencing
and Matchmaker Exchange Databases [14]. Variants in _ARHGAP35_ were named based on reference sequence NM_004491.4 and human Genome Build hg19 and evaluated according to ACMG/AMP guidelines
[15]. RESULTS Novel damaging variants in _ARHGAP35_ were identified in five individuals with developmental ocular disorders from four families. Pathogenic variants in other MAC/ASD genes
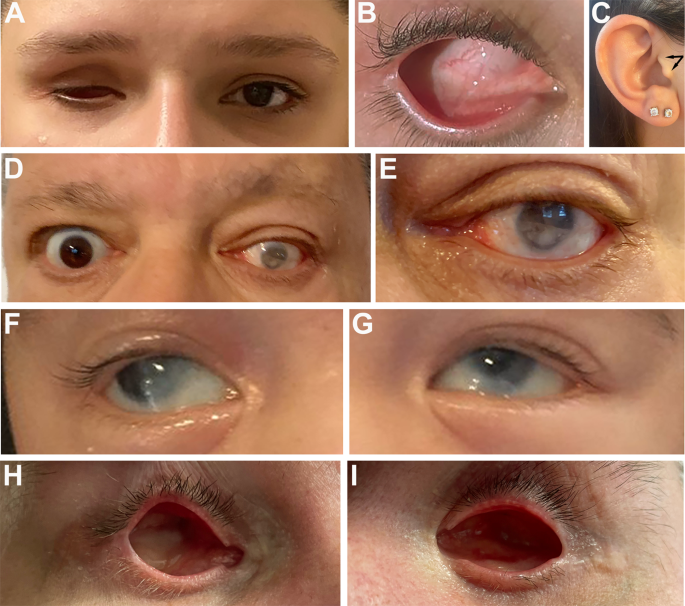
were ruled out in all individuals. Individual 1A is a 27-year-old female born with right ocular anomalies consisting of severe microphthalmia (axial length 7.6 mm) and sclerocornea; her left
eye is normal (Fig. 1A, B). She does have a bifid tragus on both ears. Her father, Individual 1B, is a 68-year-old male born with left ocular anomalies and esotropia; his right eye is
normal (Fig. 1C, D). He was diagnosed with left mild microphthalmia with iris and chorioretinal coloboma and a localized spoke shaped opacity in the lens at 6 o’clock at 6 months of age.
Additional features included a hemangioma on the right forearm and history of cancer (histocyte rich B cell non-Hodgkin’s lymphoma at 38 years of age and Nodular Lymphocyte Predominant
Hodgkin Lymphoma at 61 years of age). Growth and development were normal for both individuals. Exome sequencing identified a heterozygous novel variant in _ARHGAP35_ c.4251delC
p.(Thr1418Argfs*381) shared by both affected individuals. This variant was not present in five unaffected family members: mother, brother, paternal grandmother, and two paternal aunts
(paternal grandfather unavailable) (Table 1, Fig. 2, Supplementary Fig. 1A). This variant meets ACMG criteria to be considered Pathogenic (PVS1, PM2_supporting, PP1). Individual 2 is a
2-year-old male with bilateral type II Peters anomaly consisting of corneal opacity with cataract, iris hypoplasia, and glaucoma treated with keratoprostheses (Fig. 1E, F). Additional
features included pulmonary stenosis and thickened aortic leaflet, large left kidney with possible duplex anatomy, nevus flammeus of the glabella, small capillary hemangioma on the occiput,
and nuchal cord at birth. He has had normal growth and development to date. Trio exome sequencing of the child and unaffected parents identified a de novo novel variant in _ARHGAP35_,
c.4444delC p.(Gln1482Serfs*317) (Table 1, Fig. 2, Supplementary Fig. 1B). This variant meets ACMG criteria to be considered Pathogenic (PVS1, PS2, PM2_supporting). Individual 3 is a
3-year-old male who presented with bilateral microphthalmia (axial lengths 13.65 mm and 14.31 mm) and agenesis of the optic nerves. Sclerocornea was observed in the right eye while the left
had iris hypoplasia and corectopia. Growth was normal but development showed hypotonia and significant delay (non-ambulatory at 27 months). Additional features include left duplicated
ureters and macrocephaly (55 cm at 27 weeks, +4.55 SD). Echocardiogram, skeletal survey, and Brain MRI were normal other than eye and optic nerve anomalies. Pregnancy history is notable for
increased nuchal translucency at 12 weeks gestation and identification of renal anomalies and macrocephaly at 22 weeks gestation. Trio genome sequencing of the child and unaffected parents
identified de novo novel variants in _PTEN_, NM_000314.8:c.2 T > G p.(Met1Arg), and _ARHGAP35_, c.1849C > T p.(Arg617Ter) (Table 1, Fig. 2, Supplementary Fig. 1C). This variant meets
ACMG criteria to be considered Pathogenic (PVS1, PS2, PM2_supporting). Individual 4 is a 42-year-old male with bilateral anophthalmia. Trio exome sequencing identified a novel variant in
_ARHGAP35_, c.4294 T > C p.(Cys1432Arg), inherited from the father, who did not have a MAC phenotype but was reported to wear glasses from a young age with no further details available
(Table 1, Fig. 2, Supplementary Fig. 1D). The exome read depth was slightly skewed in the father (37%) but Sanger sequencing showed even peaks (Supplementary Fig. 1D). This missense variant
is predicted to be damaging with high CADD (32) and GERP + + (5.6) scores and a moderate REVEL score (0.439). This variant is considered a Variant of Uncertain Significance by ACMG criteria
(PM2, PP2, PP3). DISCUSSION This study presents the first evidence implicating _ARHGAP35_ in human developmental ocular phenotypes. Three variants affected the extreme C-terminus of the
protein, with two resulting in a frameshift and C-terminal extension and the other a missense change in the Rho-GAP domain. General population data in gnomAD shows significant constraint
against both loss of function and missense variants for this gene [11]. The MAC phenotypes observed here are very consistent with those reported in the mouse model. ARHGAP35 was also
previously shown to be involved in the regulation of genes within the Hippo signaling pathway [16] including _YAP1_, which is independently associated with MAC disorders [17,18,19]. Corneal
defects, observed in three families in this study, are also seen in _Yap1-_deficient mice [20]. The non-ocular features within our cohort were more variable. The presence of duplicated renal
structures in two individuals is intriguing, though it does not perfectly replicate the hypodysplasia phenotype observed in mice. With regards to the history of cancer in Individual 1B,
while a tumor suppressor role has been identified for ARHGAP35, particularly in carcinomas [5], an association with lymphoma has not been reported to date so it is unclear whether this
phenotype is coincidental or related to the genetic variant. Interestingly, only one variant was associated with an additional neurological phenotype in this cohort (Individual 3,
p.(Arg617Ter)). That variant occurred earlier in the gene, within the first exon, and is expected to lead to complete loss of function due to nonsense-mediated decay. Mice with neurological
phenotypes similarly had complete loss-of-function alleles [7, 8]. The other three variants identified in individuals with normal neurological function occurred in or after the RhoGAP domain
in the final exon of the gene; thus, the variant proteins would be expected to escape nonsense-mediated decay and may retain enough function for normal neurological development. However,
the patient with a neurological phenotype also has a _PTEN_ variant in the initiation codon. While similar _PTEN_ variants have been reported in individuals with macrocephaly and variable
autism/intellectual disability [21], the hypotonia/gross motor phenotype is more severe than is typical for _PTEN_ variants, so it is likely that the _ARHGAP35_ variant is also contributing
to the neurological phenotype. Two of the four variants were de novo, supporting a likely causative role. One of the others was inherited from an affected parent- while it could not be
conclusively proven to be de novo, it was not present in five unaffected family members. Interestingly, in this family both affected individuals had a unilateral ocular phenotype, similar to
the recently reported _PRR12_ gene with dominant unilateral MAC or ASD phenotypes [22]. In the final case, the variant was inherited from a parent without MAC but with reduced vision that
required corrective lenses. Because available clinical data was limited, it is impossible to conclude whether this represents a case of incomplete penetrance, variable expressivity, or
somatic mosaicism (though no strong evidence of mosaicism was detected); it is also possible that this is a benign allele that does not contribute to the ocular phenotype. Incomplete
penetrance and variable expressivity are noted for other MAC genes including _YAP1_ (regulated by ARHGAP35). In combination with the mouse model, this study strongly supports a role for
_ARHGAP35_ in vertebrate ocular development, consistent with its known regulation of Hippo-YAP signaling. C-terminal clustering of the identified alleles indicates a possible common
mechanism for ocular disease. Other variable developmental features were present in several patients indicating a possible role for _ARHGAP35_ in other systems, consistent with the mouse
model. Given its known role as a tumor suppressor, further investigation is needed to determine whether individuals with ocular phenotypes are at increased risk for cancer. DATA AVAILABILITY
_ARHGAP35_ variants were submitted to ClinVar (Accession numbers SCV002605061 - SCV002605064). There are no additional data available. REFERENCES * Ma A, Yousoof S, Grigg JR, Flaherty M,
Minoche AE, Cowley MJ, et al. Revealing hidden genetic diagnoses in the ocular anterior segment disorders. Genet Med. 2020;22:1623–32. Article CAS PubMed PubMed Central Google Scholar *
Chassaing N, Causse A, Vigouroux A, Delahaye A, Alessandri JL, Boespflug-Tanguy O, et al. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia.
Clin Genet. 2014;86:326–34. Article CAS PubMed Google Scholar * Patel A, Hayward JD, Tailor V, Nyanhete R, Ahlfors H, Gabriel C, et al. The Oculome Panel Test: Next-Generation Sequencing
to Diagnose a Diverse Range of Genetic Developmental Eye Disorders. Ophthalmology. 2019;126:888–907. Article PubMed Google Scholar * Jackson D, Malka S, Harding P, Palma J, Dunbar H,
Moosajee M. Molecular diagnostic challenges for non-retinal developmental eye disorders in the United Kingdom. Am J Med Genet C Semin Med Genet. 2020;184:578–89. * Heraud C, Pinault M,
Lagree V, Moreau V. p190RhoGAPs, the ARHGAP35- and ARHGAP5-Encoded Proteins, in Health and Disease. Cells 2019;8:351. * Levay M, Bartos B, Ligeti E. p190RhoGAP has cellular RacGAP activity
regulated by a polybasic region. Cell Signal. 2013;25:1388–94. Article CAS PubMed Google Scholar * Brouns MR, Matheson SF, Hu KQ, Delalle I, Caviness VS, Silver J, et al. The adhesion
signaling molecule p190 RhoGAP is required for morphogenetic processes in neural development. Development. 2000;127:4891–903. Article CAS PubMed Google Scholar * Stewart K, Gaitan Y,
Shafer ME, Aoudjit L, Hu D, Sharma R, et al. A Point Mutation in p190A RhoGAP Affects Ciliogenesis and Leads to Glomerulocystic Kidney Defects. PLoS Genet. 2016;12:e1005785. Article PubMed
PubMed Central Google Scholar * Armes JE, Williams M, Price G, Wallis T, Gallagher R, Matsika A, et al. Application of Whole Genome Sequencing Technology in the Investigation of Genetic
Causes of Fetal, Perinatal, and Early Infant Death. Pediatr Dev Pathol. 2018;21:54–67. Article PubMed Google Scholar * Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, et
al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–62. Article CAS PubMed PubMed Central Google Scholar * Karczewski KJ,
Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43. Article CAS PubMed
PubMed Central Google Scholar * Deml B, Reis LM, Lemyre E, Clark RD, Kariminejad A, Semina EV. Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur J Hum
Genet. 2016;24:535–41. Article CAS PubMed Google Scholar * Weh E, Reis LM, Happ HC, Levin AV, Wheeler PG, David KL, et al. Whole exome sequence analysis of Peters anomaly. Hum Genet.
2014;133:1497–511. Article CAS PubMed PubMed Central Google Scholar * Philippakis AA, Azzariti DR, Beltran S, Brookes AJ, Brownstein CA, Brudno M, et al. The Matchmaker Exchange: a
platform for rare disease gene discovery. Hum Mutat. 2015;36:915–21. Article PubMed PubMed Central Google Scholar * Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al.
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for
Molecular Pathology. Genet Med. 2015;17:405–24. Article PubMed PubMed Central Google Scholar * Frank SR, Kollmann CP, Luong P, Galli GG, Zou L, Bernards A, et al. p190 RhoGAP promotes
contact inhibition in epithelial cells by repressing YAP activity. J Cell Biol. 2018;217:3183–201. Article CAS PubMed PubMed Central Google Scholar * Williamson KA, Rainger J, Floyd JA,
Ansari M, Meynert A, Aldridge KV, et al. Heterozygous loss-of-function mutations in YAP1 cause both isolated and syndromic optic fissure closure defects. Am J Hum Genet. 2014;94:295–302.
Article CAS PubMed PubMed Central Google Scholar * Oatts JT, Hull S, Michaelides M, Arno G, Webster AR, Moore AT. Novel heterozygous mutation in YAP1 in a family with isolated ocular
colobomas. Ophthalmic Genet. 2017;38:281–3. Article PubMed Google Scholar * DeYoung C, Guan B, Ullah E, Blain D, Hufnagel RB, Brooks BP. De novo frameshift mutation in YAP1 associated
with bilateral uveal coloboma and microphthalmia. Ophthalmic Genet. 2022;43:513–7. Article CAS PubMed Google Scholar * Kim S, Thomasy SM, Raghunathan VK, Teixeira LBC, Moshiri A,
FitzGerald P, et al. Ocular phenotypic consequences of a single copy deletion of the Yap1 gene (Yap1 (+/-)) in mice. Mol Vis. 2019;25:129–42. PubMed PubMed Central Google Scholar *
Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, et al. The Human Gene Mutation Database (HGMD((R))): optimizing its use in a clinical diagnostic or research setting. Hum Genet.
2020;139:1197–207. Article PubMed PubMed Central Google Scholar * Reis LM, Costakos D, Wheeler PG, Bardakjian T, Schneider A, Fung SSM et al. Dominant variants in PRR12 result in
unilateral or bilateral complex microphthalmia. Clin Genet. 2021;99:437–42. Download references ACKNOWLEDGEMENTS We would like to express gratitude to the individuals and families who
participated in this study. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Pediatrics and Children’s Research Institute, Medical College of Wisconsin and Children’s Wisconsin,
Milwaukee, WI, USA Linda M. Reis, Samuel Thompson & Elena V. Semina * Service de Génétique Médicale, Hôpital Purpan CHU Toulouse, Toulouse, France Nicolas Chassaing * Platerforme
AURAGEN, Lyon, France Nicolas Chassaing * Einstein Medical Center Philadelphia, Philadelphia, PA, USA Tanya Bardakjian & Adele Schneider * Department of Ophthalmology and Visual
Sciences, Medical College of Wisconsin, Milwaukee, WI, USA Elena V. Semina Authors * Linda M. Reis View author publications You can also search for this author inPubMed Google Scholar *
Nicolas Chassaing View author publications You can also search for this author inPubMed Google Scholar * Tanya Bardakjian View author publications You can also search for this author
inPubMed Google Scholar * Samuel Thompson View author publications You can also search for this author inPubMed Google Scholar * Adele Schneider View author publications You can also search
for this author inPubMed Google Scholar * Elena V. Semina View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS LMR enrolled participants,
collected clinical data, and analyzed exome data. NC provided clinical assessment and analysis of genome data. TB was involved in enrollment and clinical data collection. ST completed Sanger
sequencing analysis. AS provided clinical assessment. EVS designed and supervised the study. LMR and EVS wrote the original draft of the paper; NC, TB, ST, and AS edited the paper.
CORRESPONDING AUTHOR Correspondence to Elena V. Semina. ETHICS DECLARATIONS FUNDING Funding for this study was provided by NIH grants EY015518 and EY025718 (EVS). COMPETING INTERESTS The
authors declare no competing interests. ETHICS APPROVAL This human study was approved by the Institutional Review Boards of Children’s Wisconsin (#124172) and Einstein Medical Center
Philadelphia (#HN2191). Written informed consent was obtained from participants and/or legal guardians for all research study activities and photo publication (if applicable). ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
FIGURE 1 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes
were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If
material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Reis, L.M., Chassaing, N., Bardakjian, T. _et al._ _ARHGAP35_ is a novel factor disrupted in human developmental eye phenotypes. _Eur J Hum Genet_ 31, 363–367 (2023).
https://doi.org/10.1038/s41431-022-01246-z Download citation * Received: 30 September 2022 * Revised: 09 November 2022 * Accepted: 15 November 2022 * Published: 01 December 2022 * Issue
Date: March 2023 * DOI: https://doi.org/10.1038/s41431-022-01246-z SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Usmca: the 3 most important changes in the new nafta and why they matterPresident Donald Trump and Democratic leaders agreed on a deal to pass a new trade agreement between the U.S., Mexico an...
England secure eurohockey spot with slovakia victoryENGLAND 11 Howard 4' PC, Peel 14' FG, Owsley 19' PC, Hamilton 20' FG, 49 FG', Martin 23' P...
Queen left ‘exhausted’ trying to ‘forge a reconciliation’The Queen was left “exhausted” trying to “forge a reconciliation” between Prince William and Harry, claimed royal author...
Bbc antiques road trip experts clash in heated 'fix' row at auctionTensions ran high in a recent episode of Antiques Road Trip as Catherine Southon squared off with Charles Hanson during ...
Page Not Found.Page Not Found. Hopefully you will find what you are looking for here....
Latests News
Arhgap35 is a novel factor disrupted in human developmental eye phenotypesABSTRACT ARHGAP35 has known roles in cell migration, invasion and division, neuronal morphogenesis, and gene/mRNA regula...
Channelnews : ifa 2018: ecovacs adds ai upgrade to cleaning botsChinese-based Ecovacs has sought to make its robotic cleaners even smarter, debuting new artificial intelligence softwar...
National hockey fund for state schools & edi | england hockeyTo make a lasting impact on hockey delivery in schools, we need to leverage existing networks and structures, building o...
Hong kong lawmakers call for more precautions to avoid spread of bedbugsAuthorities should step up efforts to prevent any possible spread of bedbugs in Hong Kong, lawmakers have warned after a...
Recent acquisitions | City of Paris Museum of Modern ArtSince 2007, more than 800 works have been donated. This has been a considerable boost to the Museum’s collections, which...