N6-methyladenosine-dependent pri-mir-17-92 maturation suppresses pten/tmem127 and promotes sensitivity to everolimus in gastric cancer
N6-methyladenosine-dependent pri-mir-17-92 maturation suppresses pten/tmem127 and promotes sensitivity to everolimus in gastric cancer"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT _N_6-methyladenosine (m6A) is the most common epigenetic RNA modification with essential roles in cancer progression. However, roles of m6A and its regulator METTL3 on non-coding
RNA in gastric cancer are unknown. In this study, we found elevated levels of m6A and METTL3 in gastric cancer. Increased METTL3 expression indicated poor outcomes of patients and high
malignancy in vitro and in vivo. Mechanically, m6A facilitated processing of pri-miR-17-92 into the miR-17-92 cluster through an m6A/DGCR8-dependent mechanism. The m6A modification that
mediated this process occurred on the A879 locus of pri-miR-17-92. The miR-17-92 cluster activated the AKT/mTOR pathway by targeting _PTEN_ or _TMEM127_. Compared with those with low levels
of METTL3, METTL3-high tumors showed preferred sensitivity to an mTOR inhibitor, everolimus. These results reveal a perspective on epigenetic regulations of non-coding RNA in gastric cancer
progression and provide a theoretical rationale for use of everolimus in the treatment of m6A/METTL3-high gastric cancer. SIMILAR CONTENT BEING VIEWED BY OTHERS METTL14-DEPENDENT MATURATION
OF PRI-MIR-17 REGULATES MITOCHONDRIAL HOMEOSTASIS AND INDUCES CHEMORESISTANCE IN COLORECTAL CANCER Article Open access 21 February 2023 _N_6-METHYLADENOSINE DEMETHYLASE ALKBH5 SUPPRESSES
MALIGNANCY OF ESOPHAGEAL CANCER BY REGULATING MICRORNA BIOGENESIS AND RAI1 EXPRESSION Article 26 July 2021 THE ROLE OF N6-METHYLADENOSINE-MODIFIED NON-CODING RNAS IN THE PATHOLOGICAL PROCESS
OF HUMAN CANCER Article Open access 18 July 2022 INTRODUCTION Gastric cancer is one of the most common malignancies and the third leading cause of cancer-related death worldwide1. With few
specific symptoms in early stages and with low rates of gastroscopy, most patients have already reached an advanced stage at the time of initial diagnosis. Even among patients who underwent
curative resection, 60% suffered recurrences and distant metastasis, with a median overall survival (mOS) of <12 months2. Moreover, gastric cancers with peritoneal metastasis respond
rarely to any treatments, leading to an extremely inferior prognosis with life expectancy <6 months3. The effect of targeted therapy is quite limited by the lack of dominant driver genes
in gastric cancer. Trastuzumab is the only target drug approved for the first-line treatment of advanced gastric cancer based on the TOGA trial, but its usage was confined in a small part of
the patients with ERBB2 amplification4. The anti-angiogenic drug bevacizumab only improves overall survival in non-Asian patients as the first-line treatment5. Up to now, the immune
checkpoint inhibitors, pembrolizumab (KEYNOTE-059 cohort 1)6 and nivolumab (ATTRACTION-02)7, are only approved for third-line and later treatment in gastric cancer, with response rates
<15%. Therefore, an in-depth investigation of the molecular mechanisms in gastric cancer oncogenesis and progression is critical to allow early diagnosis, innovative therapeutic methods,
and ultimately improved prognosis and quality of life for patients. _N_6-Methyladenosine (m6A) is the most common epigenetic modification in eukaryotic messenger RNA (mRNA)8 and non-coding
RNA (ncRNA)9. It plays a crucial role in gene expression by participating in almost every stage of mRNA metabolism and exerts vital and specific roles in the pathogenesis of various
cancers10. Recently, several studies reported that METTL3, the core methyltransferase for m6A modification, promotes gastric cancer progression11,12. However, these studies only focused on
m6A of mRNA, and few studies investigated m6A of ncRNA in gastric cancer or implied the possible clinical translational value of m6A/METTL3 related signaling pathways. Here, we investigated
the biological function of METTL3/m6A in regulating ncRNA and defined a novel pathway for m6A-dependent primary microRNA (miRNA) maturation and AKT/mTOR activation in gastric cancer, which
could be counteracted by everolimus. MATERIALS AND METHODS CELL CULTURE AND TRANSFECTION AGS, HGC-27, and MKN-45 cell lines were obtained from the Cell Bank, Chinese Academy of Science
(Shanghai, China). All cells were authenticated and tested for mycoplasma contamination. Overexpressing plasmids were transfected by Lipofectamine 2000 (Thermo Fisher Scientific, Waltham,
MA). Lentivirus containing METTL3 and shRNA against METTL3 (shM3: CCGGCGTCAGTATCTTGGGCAAGTTCTCGAGAACTTGCCCAAGATACTGACGTTTTTG and shM3-2:
CCGGGCTGCACTTCAGACGAATTATCTCGAGATAATTCGTCTGAAGTGCAGCTTTTTG) were used for stable transfection, followed by puromycin selection. The miniMIR17HG plasmid was constructed by inserting the
961–1917th nucleotides of the miR-17-92a-1 RNA (GenBank #: NR_027350.1) into the GV146 vector. CLINICAL SAMPLES Fresh tissues were obtained from gastric cancer patients who underwent radical
resections at Qilu Hospital of Shandong University between January 2017 and August 2018. Pathologically confirmed gastric cancer paraffin-embedded tissues between 2009 and 2014 were
obtained from the Department of Pathology, Qilu Hospital of Shandong University. Only patients with evidence of survival and recurrence were included for OS and RFS analysis, respectively.
RNA M6A QUANTIFICATION Total RNA was isolated from 10 mm3 of fresh tissue or 106 cells using TRIzol (Invitrogen, CA, USA). The m6A RNA Methylation Quantification Kit (Abcam, Cambridge, UK)
was used to quantify the m6A content according to the manufacturer’s instructions. The optical absorbance was measured by a SpectraMax Plus384 Microplate Spectrophotometer (Molecular Device,
Sunnyvale, CA, USA). IMMUNOHISTOCHEMISTRY (IHC) In clinical studies, paraffin-embedded sections were blocked by goat serum and stained with anti-METTL3 antibody (Abcam, Cambridge, UK) using
an IHC staining kit (Zsbio, Beijing, China). Cell nuclei were stained with hematoxylin. In animal studies, tumor xenografts or peritoneal tumors were fixed and processed with a similar
procedure with anti-METTL3, anti-Ki67 (Abcam, Cambridge, UK), and anti-PTEN antibody (CST, Danvers, MA). PROLIFERATION AND COLONY-FORMATION ASSAYS For proliferation assays, cells were seeded
in 6-well plates (10,000 or 20,000 cells per well) and counted every 24 h. For colony-formation assays, suspended single cells were seeded in 6-well plates (1000 cells per well), and
colonies were counted within 14 days. WOUND-HEALING, MIGRATION, AND INVASION ASSAYS For wound-healing assays, wounds were made by scratching a line using a 200 µL tip, and the intervals were
measured within 72 h. For migration and invasion assays, a Transwell system (Corning, NY, USA) was used as previously described13. Migrated and invaded cells were stained with crystal
violet (Beyotime, Shanghai, China) and photographed. SUBCUTANEOUS XENOGRAFT AND PERITONEAL IMPLANT MODELS Six-week female BALB/c Nude Mice were purchased from Vital River Laboratory
(Beijing, China). For subcutaneous xenograft models, 0.1 mL of cell suspension containing 106 cells were injected subcutaneously into the right flank of mice (_n_ = 6 for each group). Mice
were sacrificed at 21- or 32-day after injection. For peritoneal implant models, a cell suspension (5 × 106 cells) was injected intraperitoneally. All mice were sacrificed 4 weeks after
injection (_n_ = 4 for each group). Mice were randomly allocated into each group with no blinding. The animal studies were performed following the ARRIVE guidelines and were approved by the
Animal Ethical Committee of Qilu Hospital of Shandong University. WESTERN BLOT Approximately 25 µg of protein was separated by 10% SDS-PAGE, transferred to 0.22 μm polyvinylidene difluoride
membranes (Thermo Fisher Scientific), and probed by primary antibodies. The anti-METTL3, anti-TMEM127, and anti-GAPDH antibodies were purchased from Abcam (Cambridge, UK). The anti-PTEN,
anti-phospho-S6K, anti-phospho-S6, and anti-phospho-4E-BP1 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). RNA IMMUNOPRECIPITATION (RIP) A Magna RIP RNA-Binding
Protein Immunoprecipitation Kit (Millipore, Darmstadt, Germany) was used for RIP. Briefly, cells were lysed and mixed with anti-m6A (Abcam, Cambridge, UK), anti-DGCR8 (Abcam, Cambridge, UK)
antibodies, or isotype controls (Abcam, Cambridge, UK). The antibody-binding RNA was pulled down by protein A/G magnetic beads and quantified by real-time PCR. QUANTITATIVE RT-PCR
Quantification of miRNA was performed using an All-in-One™ miRNA qRT-PCR Detection Kit (GeneCopoeia, San Diego, CA, USA). Quantification of primary miRNA and mRNA was performed using an
All-in-One™ First-Strand cDNA Synthesis Kit and an All-in-One qPCR Kit (GeneCopoeia, San Diego, CA, USA). Commercial primers for quantitation of miR-17, miR-18, miR-19a, miR-19b-1, miR-20a,
miR-92a-1, snRNA U6, METTL3, TMEM127, PTEN, and GAPDH were purchased from GeneCopoeia (San Diego, CA, USA). Primers for pri-miR-17-92 included 5′-CATCTACTGCCCTAAGTGCTCCTT and
5′-GCTTGGCTTGAATTATTGGATGA. Primers for 5S RNA were 5′-TCTCGTCTGATCTCGGAAGC and 5′-AGCCTACAGCACCCGGTATT. EVEROLIMUS-SENSITIVITY ASSAYS For the in vitro study, everolimus (APExBIO, Houston,
TX, USA) was added to cells with final concentrations of 5 or 50 µg/mL. Cell viability was measured using a CCK-8 kit (BestBio, Shanghai, China). For the in vivo study, subcutaneous
xenograft models with METTL3-high (_n_ = 3) and control cells (_n_ = 3) were established as above. Control cells were inoculated two days before METTL3-high cells. When the tumor sizes were
similar, volume-matched mice received everolimus (50 µg/day intragastrically) or solvent for 17 days and were sacrificed at day 18. Mice were randomly allocated to each group with no
blinding. IN SILICO ANALYSES All datasets used in this study were derived from public databases. Enrichment analyses were performed by DAVID (https://david.ncifcrf.gov) and TAM
(http://www.cuilab.cn/tam). Prediction of miRNA targets was performed by the online tool miRDB (http://mirdb.org/). STATISTICAL ANALYSES Data were from at least three independent experiments
unless otherwise specified. Variations within each group were estimated and were all statistically compared. Differences between groups were calculated by Student’s _t_-tests. Univariate
analyses were performed by Chi-squared tests. Survival data were compared by log-rank tests. Correlations were analyzed by linear regression. All the statistical tests were two-tailed. _P_
values < 0.05 were considered statistically significant. Data were presented as mean or mean ± standard deviation (SD). RESULTS ELEVATED M6A IS MAINLY REGULATED BY ITS “WRITER” METTL3 IN
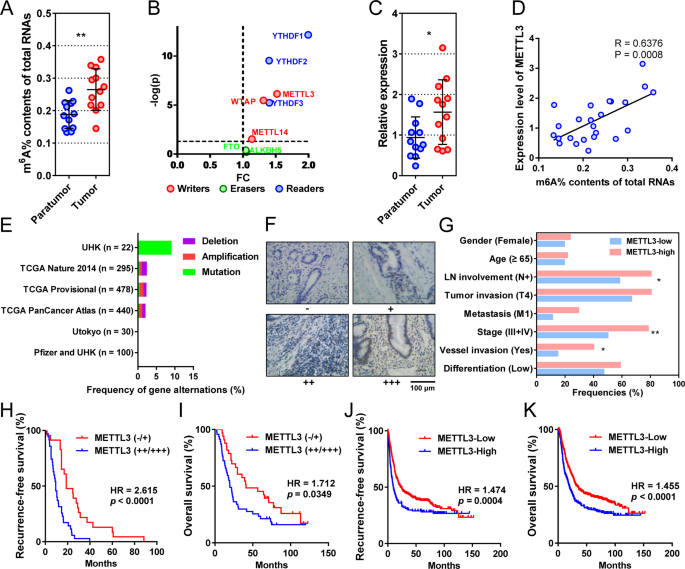
GASTRIC CANCER To explore the features of m6A in gastric cancer, we first compared the levels of m6A on total RNAs from 12 pairs of cancerous and adjacent tissues. The m6A levels were
significantly elevated in tumor tissues compared with their adjacent tissues (Fig. 1a). To determine the key regulators, we performed differential expression analysis on the m6A “writers”,
“erasers”, and “readers” from gastric cancer and normal tissues in The Cancer Genome Atlas Stomach Adenocarcinoma (TCGA-STAD) database. The “writers” and “readers” were all overexpressed in
gastric cancer, whereas “erasers” were expressed similarly in tumors and controls (Fig. 1b). Based on this, we then focused on the most differentially expressed “writer”, METTL3, for further
study. METTL3 RNA level was significantly higher in cancerous tissues than that in adjacent tissues (Fig. 1c). In addition, METTL3 RNA positively correlated with total RNA m6A levels in our
cohort (Fig. 1d). When mutations were considered in the previously published cohorts, genetic changes in METTL3 only accounted for 2.12% of total patients (Fig. 1e). METTL3 OVEREXPRESSION
INDICATES ADVERSE PATHOLOGICAL FEATURES AND POOR OUTCOME IN GASTRIC CANCER To determine the clinical relevance of METTL3, we tested METTL3 expression in gastric cancer samples of 87 patients
who had received radical or palliative gastrectomy (Fig. 1f). METTL3 was mainly distributed in the nucleus of tumor cells. According to the staining intensity, 12 negative (−), 24 weak
positive (+), 32 medium positive (++), and 19 strong positive cases (+++) were observed. METTL3 expression significantly correlated with AJCC staging (the eighth edition, _P_ = 0.0056),
lymph node metastasis (_P_ = 0.0251), and vascular invasion (_P_ = 0.0346), but did not correlate with gender, age, differentiation, and tumor invasion (Table 1, Fig. 1g). Patients with
METTL3 overexpression (++/+++) had higher risks of recurrence (hazard ratio (HR) = 2.615, 95% confidence interval (CI) = 1.934–5.623, Fig. 1h) and death (HR = 1.712, 95% CI = 1.042–2.799,
Fig. 1i), with significantly shorter median recurrence-free survival (mRFS, 9.3 vs. 18.7 months, _P_ = 0.0002, Fig. 1h) and mOS (19.6 vs. 41.1 months, _P_ = 0.0349, Fig. 1i) than those with
low METTL3 expression (−/+). In addition, the prognostic value of METTL3 was also confirmed by the online tool Kaplan–Meier plotter. In this database, the METTL3-high patients had higher
risks of recurrence (HR = 1.474, 95% CI = 1.211–1.955, Fig. 1j) and death (HR = 1.455, 95% CI = 1.238–1.841, Fig. 1k), with shorter mRFS (9.7 vs. 19.8 months, _P_ = 0.0004, Fig. 1j) and mOS
(19.8 vs. 33.2 months, _P_ < 0.0001, Fig. 1k) than the METTL3-low gastric cancer patients. METTL3 PROMOTES GASTRIC CANCER CELL PROLIFERATION AND TUMOR GROWTH To investigate the biological
role of METTL3 in gastric cancer, we established METTL3-downregulated (Fig. 2a and supplementary Fig. S1a) and -upregulated cells (Fig. 2b) using lentivirus containing shRNA and plasmids
containing METTL3, respectively. Compared to those in control cells, the m6A levels of total RNA were significantly decreased in METTL3-downregulated cells (_P_ = 0.0024 and 0.0015, Fig. 2c
and supplementary Fig. S1b) and elevated in METTL3-upregulated cells (_P_ = 0.0082 and 0.0002, Fig. 2d). In cell models, METTL3 downregulation impeded cell proliferation (all _P_ < 0.05
from day 2, Fig. 2e and supplementary Fig. S1c) and colony formation (_P_ = 0.0004 and 0.0010, Fig. 2f and supplementary Fig. S1d). Accordingly, METTL3 overexpression stimulated cell
proliferation (all _P_ < 0.05 from day 3, Fig. 2g) and colony-forming efficiencies (_P_ = 0.0002 and 0.0055, Fig. 2h). Subsequently, subcutaneous xenograft models were established for in
vivo studies. The median times to form palpable tumors were 11.0 and 13.5 days in METTL3-overexpressed and control cells, respectively (_P_ = 0.0055, Fig. 2i, j). Three weeks after
subcutaneous injection, tumors from METTL3-overexpressing cells were remarkably larger (_P_ = 0.0028, Fig. 2k, l) and heavier (_P_ = 0.0027, Fig. 2l) than tumors from control cells. In
addition, Ki67 was also overexpressed in METTL3-high tumors (_P_ < 0.0001, Fig. 2m). In mice with METTL3-low tumors, the median time to form touchable tumors was significantly longer than
that in control mice (24.0 vs. 22.0 days, _P_ = 0.0055, Fig. 2i, n). Further, METTL3-low tumors were significantly smaller (_P_ = 0.0369, Fig. 2o, p) and had lower weights (_P_ = 0.0421,
Fig. 2p) and Ki67 expression (_P_ = 0.0005, Fig. 2q) than the control group. METTL3 PROMOTES GASTRIC CANCER CELL MIGRATION, INVASION, AND PERITONEAL METASTASIS In wound-healing assays,
METTL3 downregulation reduced migration distances remarkably (_P_ = 0.0023 and 0.0005, Fig. 3a and supplementary Fig. S1e), while METTL3 overexpression increased the migration distances (_P_
< 0.0001 and =0.0003, Fig. 3b). In Transwell assays, the METTL3-low cells showed decreased migration (_P_ = 0.0199 and 0.0061, Fig. 3c and supplementary Fig. S1f) and invasion (_P_ =
0.0029 and 0.0003, Fig. 3d and supplementary Fig. S1g) compared to the control cells. Accordingly, METTL3 overexpression significantly augmented cell migration (_P_ = 0.0001 and 0.0010, Fig.
3e) and invasion (_P_ < 0.0001 and =0.0028, Fig. 3f) in two cell models. Next, we established a gastric cancer peritoneal metastasis model by intraperitoneal injection of tumor cells
(Fig. 3g). The abdominal circumferences of the mice bearing METTL3-high cells increased rapidly and became larger than those of the mice bearing control cells from the second week (all _P_
< 0.05, Fig. 3h). However, body weights were not significantly different between the two groups (Fig. 3i). Most mice with METTL3-high cells showed palpable masses on the abdominal wall,
which was not observed in the control group (Fig. 3g). When dissected four weeks later, the mice bearing METTL3-high cells possessed more and larger peritoneal-implanted nodules (both _P_
< 0.0001, Fig. 3j, k). They were mainly distributed in the mesentery and omentum (Fig. 3j), while few grew on the surface of the liver or spleen. METTL3 overexpression in implanted
peritoneal nodes was confirmed by IHC (Fig. 3l). METTL3 PROMOTES TUMOR PROGRESSION BY FACILITATING BIOGENESIS OF MIR-17-92 CLUSTER To decipher the mechanisms of METTL3 in tumor growth and
metastasis, we performed correlation analysis between METTL3 mRNA and all the other mRNA, miRNA, and long ncRNA in the TCGA-STAD database. Among the top 7 METTL3-correlated miRNAs, 6
(miR-17, 19b-1, 20a, 92a-1, 19a, and 18a) were derived from one miRNA cluster, the miR-17-92 cluster, which shared a primary miRNA, the pri-miR-17-92 (the transcript of _MIR17HG_, Fig. 4a,
b). Meanwhile, none of these miRNAs targeted METTL3 mRNA, as predicted by miRDB. By cluster enrichment analysis of miRNA with software TAM, 76 METTL3-correlated miRNAs (correlation
coefficients >0.2 and FDR < 4 × 10−6) were also significantly enriched in the miR-17-92 cluster (_P_ < 0.0001, Fig. 4c). In terms of function, these METTL3-correlated miRNAs were
enriched in several cancer-associated pathways, such as cell proliferation, immunity, AKT pathway, onco-miRNA, angiogenesis, apoptosis, and cell cycle (Fig. 4d). We then examined the
expression levels of the miR-17-92 cluster and pri-miR-17-92 in cells and xenograft tumors. Both in vitro or in vivo, METTL3 overexpression significantly increased levels of all six miRNAs
(all _P_ < 0.05, Fig. 4e) and reduced the level of pri-miR-17-92 (_P_ = 0.0229 in vitro and _P_ = 0.0009 in vivo, Fig. 4f), while METTL3 downregulation reduced all miRNAs (all _P_ <
0.05, Fig. 4g) and increased the level of pri-miR-17-92 (_P_ = 0.0078 in vitro and _P_ = 0.0009 in vivo, Fig. 4h). The correlation between METTL3 and the miRNA-17-92 cluster was also
confirmed in our clinical samples. Levels of miR-17, 18, 19a, 19b-1, and 20a, as well as the average level of all six miRNAs (miR-mean), were significantly elevated in tumors compared with
those in adjacent tissues (_P_ < 0.05 for all, Fig. 4i). The expression of each member of the miRNA-17-92 cluster was positively correlated with the others (Fig. 4j). In addition, most
members (except miR-92a-1) and miR-mean correlated positively with the level of METTL3 mRNA with statistical significance (_P_ < 0.05 for all, Fig. 4j, k). To verify whether METTL3 exerts
the onco-promoting role by facilitating pri-miR-17-92 maturation, we constructed a miniMIR17HG plasmid to force expression of 6 miRNAs for rescue studies14. This construct contains all
miRNAs from the miR-17-92 cluster but no flanking sequences and can be rapidly processed into mature miRNAs. Transfection of miniMIR17HG in METTL3-knocked-down cells significantly raised all
miRNAs (all _P_ < 0.01) to similar levels of control cells (Fig. 4l). In addition, miniMIR17HG partially rescued cell proliferation (_P_ < 0.05 from day 2, Fig. 4m) and cell
migrations (_P_ = 0.0019, Fig. 4n). METTL3 FACILITATES PRI-MIR-17-92 PROCESSING BY M6A MODIFICATION ON ITS A879 LOCUS To explore the mechanisms by which METTL3 regulates pri-miR-17-92
processing, we examined its function on m6A regulation of pri-miR-17-92 and pri-miR-17-92-DGCR8 binding by RNA RIP. METTL3 overexpression significantly increased the m6A modification on
pri-miR-17-92 (_P_ < 0.0001, Fig. 5a). Besides, METTL3 remarkably facilitated DGCR8 binding to pri-miR-17-92 (_P_ = 0.0077, Fig. 5a). Consistently, METTL3 downregulation repressed m6A
modification on pri-miR-17-92 (_P_ = 0.0021) and DGCR8-pri-miR-17-92 binding (_P_ < 0.0001, Fig. 5b). To identify the specific sites of m6A on pri-miR-17-92 by METTL3, we analyzed its
sequence and found five “GGAC” motifs that were potentially recognized by METTL3 (Fig. 5c). Five plasmids with adenine to cytosine mutation in these motifs were constructed and transfected
into cells (Fig. 5c). Compared to the wild type plasmid, A879C mutation hindered miRNA biogenesis significantly, while the others did not (Fig. 5d). Also, A879C mutation remarkably reduced
the precipitated pri-miR-17-92 by anti-m6A antibody in control cells with normal expression of METTL3 (_P_ = 0.0004), but this difference was not observed in METTL3-knocked-down cells (Fig.
5e). In another aspect, the wild type pri-miR-17-92 with m6A in control cells was 4.3 times as much as that in METTL3-low cells (Fig. 5f). In contrast, the difference of pri-miR-17-92-A879C
with m6A was only 1.5 times in these two cell lines, indicating a key role of A879 in the METTL3-mediated pri-miR-17-92 processing (Fig. 5f). In cell proliferation assays, cells transfected
with wild type pri-miR-17-92 showed superior proliferation ability to both pri-miR-17-92-A879C-expressing and control cells from day 1 to 4 (all _P_ < 0.05 from day 2, Fig. 5g). In
addition, cells transfected with pri-miR-17-92-A879C also showed inferior migrating ability compared to those transfected with wild type pri-miR-17-92 (_P_ = 0.0082, Fig. 5h). METTL3
INHIBITS PTEN/TMEM127 EXPRESSION AND ACTIVATES AKT/MTOR PATHWAY BY FACILITATING BIOGENESIS OF MIR-17-92 CLUSTER To determine the downstream targets of the miR-17-92 cluster in gastric cancer
progression, we performed miRNA cluster enrichment analysis and found the members of the miR-17-92 cluster were significantly enriched in several cancer-related terms, including cell
proliferation, AKT pathway, angiogenesis, onco-miRNAs, and apoptosis (Fig. 6a). Also, we obtained 2056 target genes of the miR-17-92 cluster predicted by miRDB and 525 mRNAs that negatively
correlated with METTL3 with a correlation coefficient < −0.18 in the TCGA-STAD database. The two gene pools shared a total of 98 genes, which were enriched in PI3K/mTOR, p53, TGF-β, and
MAPK pathways by KEGG analysis (Fig. 6b). Thus, we chose the members that were clustered in the PI3K/mTOR pathway, PTEN and TMEM127, for further study. In the TCGA-STAD databases, both PTEN
and TMEM127 were negatively correlated with METTL3 expression with statistical significance (both _P_ < 0.0001, Fig. 6c). These negative correlations between METTL3 mRNA and PTEN/TMEM127
mRNA were confirmed in our clinical samples by quantitative RT-PCR (Fig. 6d). Nonsynonymous mutation and copy number variations (CNVs) of _PTEN_ occurred in about 10.6% of gastric patients
(Fig. 6e). Among them, all CNVs of _PTEN_ were deletion instead of amplification (Fig. 6e). In contrast, nonsynonymous mutations and CNVs of _TMEM127_ only occurred in 1.6% of the patients,
and all CNVs were amplification (Fig. 6e). By RT-PCR, METTL3 overexpression significantly reduced mRNA levels for PTEN (_P_ < 0.0001) and TMEM127 (_P_ = 0.0019, Fig. 7a), while METTL3
downregulation elevated their mRNA levels (_P_ = 0.0002 and 0.0083, Fig. 7b). Furthermore, forced expression of miniMIR17HG in METTL3-reducing cells reversed (of PTEN, _P_ = 0.0124) or
diminished (of TMEM127, _P_ = 0.0929) the differences (Fig. 7b). By western blot and IHC, both PTEN and TMEM127 were remarkably reduced in METTL3-high cells, subcutaneous xenografts, and
peritoneal implants (Fig. 7c, g). Accordingly, when METTL3 was reduced, PTEN and TMEM127 were significantly elevated in cells and subcutaneous xenografts (Fig. 7h–j). What is more, the
overexpression of PTEN and TMEM127 caused by METTL3-knockdown was reversed by miniMIR17HG (Fig. 7k). We continued to investigate whether METTL3/miR-17-92 cluster influenced the AKT/mTOR
pathway activation. In METTL3-high cells, subcutaneous xenografts, and peritoneal metastatic nodes, the phosphorylation levels of AKT/mTOR pathway-related proteins, including AKT, S6K, S6,
and 4E-BP1, were all upregulated (Fig. 7c, d, f), which indicated the activation of the AKT/mTOR pathway. In the METTL3-reducing cells and xenograft tumors, phosphorylation levels of AKT,
S6K, S6, and 4E-BP1 were significantly decreased (Fig. 7h, i). Further, forced expression of miniMIR17HG counteracted the AKT/mTOR pathway inactivation by METTL3-knocked-down (Fig. 7k).
GASTRIC CANCER WITH METTL3 OVEREXPRESSION IS MORE SENSITIVE TO EVEROLIMUS Due to the remarkable impact of the METTL3/miR-17-92 cluster on the AKT/mTOR pathway, we explored whether
everolimus, an mTOR inhibitor, could inhibit the onco-promoting role of METTL3. Cell viability was measured in cell models with different levels of METTL3 after treatment with solvent or
different concentrations of everolimus. Without everolimus, METTL3 overexpression increased cell viability (_P_ < 0.05 from day 1, Fig. 8a), whereas METTL3 downregulation decreased cell
viability (_P_ < 0.05 from day 1, Fig. 8b), compared to their corresponding controls. Addition of everolimus remarkably suppressed cell viability, regardless of its concentrations or
METTL3 expression (Fig. 8a, b). However, METTL3-overexpressing cells showed more sensitivity to everolimus in a dose-dependent manner than the control cells (_P_ < 0.05 at day 2 and 3,
Fig. 8a). In accordance, the METTL3-low cells showed lower sensitivity to everolimus in both concentrations than the control cells (_P_ < 0.05 at day 2 and 3, Fig. 8b). With in vivo
models, everolimus was effective in reducing tumor volumes in both METTL3-overexpressing or control tumors (Fig. 8c). The tumor inhibition rate of everolimus in METTL3-high tumors was
significantly higher than that in control tumors (89.92% vs. 73.26%, _P_ = 0.0465, Fig. 8c–e), suggesting that METTL3-high tumors could be inhibited by everolimus more effectively.
DISCUSSION Due to its heterogeneity and specificity, gastric cancer has been gradually shown to lack common driven mutations or CNVs that are commonly seen in lung, breast, or colorectal
cancer. Consequently, it is difficult to copy the success of the translational therapies based on genetic alterations in certain types of cancers, such as the EGFR or ALK-driven non-small
cell lung cancer. Increasing scholars believe that cancer is an epigenetic disease, and disrupted and unstable epigenomes exist widely among different types of tumors with heterogeneity and
therapeutic resistance15,16. m6A is the most abundant internal modification in eukaryotic RNA and may represent a critical mechanism for regulating malignant behaviors of tumors17. Our data
show that the m6A level was upregulated in gastric cancers and suppressing m6A by METTL3-knockdown hindered tumor growth and metastasis in vitro and in vivo. Based on these discoveries, we
believe that abnormal m6A modification is an important epigenetic feature of gastric cancer and a potential therapeutic target for further study. As the predominant “writer” of m6A, METTL3
is dysregulated and plays dual roles in cancers, coupled with different substrates and cell types18. It directly regulates transcription, translation, and RNA maturation of a broad range of
oncogene and onco-suppressors, many of which are undiscovered, and the role of METTL3 cancers depends on orchestration of multiple effects18. In non-tumor cells, METTL3 methylated primary
miRNAs, such as pri-let-7e and pri-miR221, and facilitated their maturation19. Recent studies reported that METTL3 accelerated the maturation of pri-miR221/22220 and pri-miR-2521 in bladder
and pancreatic cancers. So far, there has been no study reported the regulating function of METTL3 on ncRNA in gastric cancer. As we found, one of the most prominent substrates of METTL3 is
a non-coding primary miRNA, pri-miR-17-92, a 6-tandem stem hairpin-containing polycistronic transcript of the gene _MIR17HG_ that encodes a miRNA cluster composed of 6 onco-miRNAs22. Through
m6A modification, METTL3 facilitated pri-miR-17-92 to bind to DGCR8, which recognizes the stem-flanking junctions and orients DROSHA to cleave primary miRNA into precursor miRNA23,24. Our
study revealed that the over-m6A modification on pri-miR-17-92, instead of its overexpression, caused upregulation of the miR-17-92 cluster and gastric cancer progression. To our knowledge,
this is the first report of METTL3-mediated-m6A-dependent maturation of ncRNA and its downstream signal pathway and biological effect in gastric cancer. Most importantly, we mapped
pri-miR-17-92 and found its A879 was the dominant site responsible for the m6A-mediated pri-miR-17-92 maturation because A879C mutation significantly abolished the METTL3-mediated m6A
modification, DGCR8 binding, and miR-17-92 cluster formation. Therefore, the pri-miR-17-92 A879 represented the key mediator of the m6A-mediated pri-miR-17-92 maturation and was likely a
highly precise target for intervention as an epigenetically modifiable position. The regulation of the miR-17-92 cluster remains largely unclear, other than that c-Myc directly binds to
_MIR17HG_ and promotes pri-miR-17-92 transcription25. Our findings uncovered another layer of regulation of this miRNA cluster by m6A-mediated processing. Widely accepted as an onco-miRNA26,
the miR-17-92 cluster targets many onco-suppressing genes, such as SMAD2 and SMAD427, p2128, and TRAF329. In this study, we found that TMEM127 and PTEN were part of the targets.
Furthermore, forced expression of the miR-17-92 cluster counteracted the effects of METTL3-knockdown in PTEN/TMEM127 regulation, cell proliferation, and metastasis. TMEM127 is a negative
regulator of the mTOR pathway and a tumor suppressor located on chromosome 2q1130. So far, the function of TMEM127 in gastric cancer and its association with the miR-17-92 cluster have not
been reported. In this study, we did not observe frequent mutations or deletions of TMEM127. Instead, TMEM127, along with PTEN and the mTOR pathway, was efficiently regulated by the
miR-17-92 cluster as its target. Therefore, we suspect that METTL3/miR-17-92 cluster activates mTOR pathways by targeting PTEN and TMEM127 in gastric cancer progression. Of note, several
other signaling pathways, including p53, TGF-beta, and MAPK signaling pathways, were probably also involved, which are of value for further investigation in future studies. Currently, drugs
targeting epigenetic changes, such as DNA methyltransferase inhibitors decitabine, have become standard treatments in hematological malignancies31. Up to now, the small molecule inhibitor
against METTL3 is not available. Based on the above discovery, the mTOR inhibitor everolimus was chosen to interfere with METTL3/miR-17-92 cluster/TMEM127 or PTEN/mTOR signaling pathway in
gastric cancer. We found that everolimus indeed reversed the METTL3-induced proliferation in a dose-dependent manner. This effect was remarkably pronounced when METTL3 was highly expressed.
The high sensitivity to everolimus in METTL3-high cells could be because the mTOR pathway was greatly activated by METTL3 in these cells. These data confirmed that the oncogenic role of
METTL3 relies on mTOR activation in another aspect. Everolimus has shown promising efficacy in patients with previously treated advanced gastric cancer in a phase II study32. However, phase
III studies33,34,35 failed to repeat the positive results, except for that the progression-free survival was significantly prolonged. Our findings provided evidence that the METTL3 level
might be a potential predictor for treatment efficacy of everolimus in gastric cancer and may help screen possible advantageous subgroups out from the previous failed clinical trials with
everolimus-treated gastric cancer. These inferences are yet to be further studied and confirmed in clinical trials. In conclusion, METTL3/m6A promotes gastric cancer growth and metastasis by
facilitating pri-miR-17-92 processing into the oncogenic miRNA cluster and activating the AKT/mTOR pathway by targeting PTEN and TMEM127, which could be targeted by everolimus (Fig. 8f).
These findings provide us with a novel insight into the role of METTL3 in the regulation of cancer development and a theoretical rationale for use of everolimus in the treatment of
m6A/METTL3-high gastric cancer. DATA AVAILABILITY Expression data of mRNA and miRNA in TCGA-STAD were downloaded from the GDC data portal (https://portal.gdc.cancer.gov). Survival data from
GEO databases, including GSE14210, GSE15459, GSE22377, GSE29272, GSE38749, GSE51105, and GSE62254, were extracted from Kaplan–Meier Plotter (http://kmplot.com). Genomic alternation data were
extracted from cBioPortal (http://www.cbioportal.org). All the other data and materials are available from the corresponding author upon reasonable request. REFERENCES * Bray, F. et al.
Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. _CA_ 68, 394–424 (2018). PubMed Google Scholar * Van Cutsem, E.,
Sagaert, X., Topal, B., Haustermans, K. & Prenen, H. Gastric cancer. _Lancet_ 388, 2654–2664 (2016). Article PubMed Google Scholar * Thomassen, I. et al. Peritoneal carcinomatosis of
gastric origin: a population-based study on incidence, survival and risk factors. _Int. J. Cancer_ 134, 622–628 (2014). Article CAS PubMed Google Scholar * Bang, Y. J. et al. Trastuzumab
in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised
controlled trial. _Lancet_ 376, 687–697 (2010). Article CAS PubMed Google Scholar * Ohtsu, A. et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced
gastric cancer: a randomized, double-blind, placebo-controlled phase III study. _J. Clin. Oncol._ 29, 3968–3976 (2011). Article CAS PubMed Google Scholar * Fuchs, C. S. et al. Safety and
efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. _JAMA Oncol._ 4, e180013
(2018). Article PubMed PubMed Central Google Scholar * Kang, Y. K. et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant
of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. _Lancet_ 390, 2461–2471 (2017). Article CAS
PubMed Google Scholar * Zhao, B. S., Roundtree, I. A. & He, C. Post-transcriptional gene regulation by mRNA modifications. _Nat. Rev. Mol. Cell Biol._ 18, 31–42 (2017). Article CAS
PubMed Google Scholar * Yang, D. et al. N6-Methyladenosine modification of lincRNA 1281 is critically required for mESC differentiation potential. _Nucleic Acids Res._ 46, 3906–3920
(2018). Article CAS PubMed PubMed Central Google Scholar * Liu, N. & Pan, T. N6-methyladenosine-encoded epitranscriptomics. _Nat. Struct. Mol. Biol._ 23, 98–102 (2016). Article CAS
PubMed Google Scholar * Wang, Q. et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. _Gut_,
https://doi.org/10.1136/gutjnl-2019-319639 (2019). * Yang, D. D. et al. METTL3 promotes the progression of gastric cancer via targeting the MYC pathway. _Front Oncol._ 10, 115 (2020).
Article PubMed PubMed Central Google Scholar * Zhang, D. et al. LIMD1 is a survival prognostic marker of gastric cancer and hinders tumor progression by suppressing activation of YAP1.
_Cancer Manag Res._ 10, 4349–4361 (2018). Article CAS PubMed PubMed Central Google Scholar * Olive, V. et al. A component of the mir-17-92 polycistronic oncomir promotes
oncogene-dependent apoptosis. _eLife_ 2, e00822 (2013). Article PubMed PubMed Central Google Scholar * Feinberg, A. P., Koldobskiy, M. A. & Gondor, A. Epigenetic modulators,
modifiers and mediators in cancer aetiology and progression. _Nat. Rev. Genet._ 17, 284–299 (2016). Article CAS PubMed PubMed Central Google Scholar * Heyn, H. et al. Epigenomic
analysis detects aberrant super-enhancer DNA methylation in human cancer. _Genome Biol._ 17, 11 (2016). Article PubMed PubMed Central Google Scholar * Roundtree, I. A., Evans, M. E.,
Pan, T. & He, C. Dynamic RNA modifications in gene expression regulation. _Cell_ 169, 1187–1200 (2017). Article CAS PubMed PubMed Central Google Scholar * Zheng, W. et al. Multiple
functions and mechanisms underlying the role of METTL3 in human cancers. _Front Oncol._ 9, 1403 (2019). Article PubMed PubMed Central Google Scholar * Alarcon, C. R., Lee, H., Goodarzi,
H., Halberg, N. & Tavazoie, S. F. N6-methyladenosine marks primary microRNAs for processing. _Nature_ 519, 482–485 (2015). Article CAS PubMed PubMed Central Google Scholar * Han, J.
et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. _Mol. Cancer_ 18, 110 (2019). Article PubMed PubMed Central
Google Scholar * Zhang, J. et al. Excessive miR-25-3p maturation via N(6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. _Nat. Commun._ 10, 1858
(2019). Article PubMed PubMed Central Google Scholar * Ota, A. et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant
lymphoma. _Cancer Res._ 64, 3087–3095 (2004). Article CAS PubMed Google Scholar * Gregory, R. I. et al. The Microprocessor complex mediates the genesis of microRNAs. _Nature_ 432,
235–240 (2004). Article CAS PubMed Google Scholar * Denli, A. M., Tops, B. B., Plasterk, R. H., Ketting, R. F. & Hannon, G. J. Processing of primary microRNAs by the Microprocessor
complex. _Nature_ 432, 231–235 (2004). Article CAS PubMed Google Scholar * O’Donnell, K. A., Wentzel, E. A., Zeller, K. I., Dang, C. V. & Mendell, J. T. c-Myc-regulated microRNAs
modulate E2F1 expression. _Nature_ 435, 839–843 (2005). Article PubMed Google Scholar * Ventura, A. et al. Targeted deletion reveals essential and overlapping functions of the miR-17
through 92 family of miRNA clusters. _Cell_ 132, 875–886 (2008). Article CAS PubMed PubMed Central Google Scholar * Mestdagh, P. et al. The miR-17-92 microRNA cluster regulates multiple
components of the TGF-beta pathway in neuroblastoma. _Mol. Cell_ 40, 762–773 (2010). Article CAS PubMed PubMed Central Google Scholar * Fontana, L. et al. Antagomir-17-5p abolishes the
growth of therapy-resistant neuroblastoma through p21 and BIM. _PloS ONE_ 3, e2236 (2008). Article PubMed PubMed Central Google Scholar * Liu, F., Cheng, L., Xu, J., Guo, F. & Chen,
W. miR-17-92 functions as an oncogene and modulates NF-kappaB signaling by targeting TRAF3 in MGC-803 human gastric cancer cells. _Int. J. Oncol._ 53, 2241–2257 (2018). CAS PubMed Google
Scholar * Qin, Y. et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. _Nat. Genet._ 42, 229–233 (2010). Article CAS PubMed PubMed Central Google Scholar *
Nie, J., Liu, L., Li, X. & Han, W. Decitabine, a new star in epigenetic therapy: the clinical application and biological mechanism in solid tumors. _Cancer Lett._ 354, 12–20 (2014).
Article CAS PubMed Google Scholar * Doi, T. et al. Multicenter phase II study of everolimus in patients with previously treated metastatic gastric cancer. _J. Clin. Oncol._ 28, 1904–1910
(2010). Article CAS PubMed Google Scholar * Ohtsu, A. et al. Everolimus for previously treated advanced gastric cancer: results of the randomized, double-blind, phase III GRANITE-1
study. _J. Clin. Oncol._ 31, 3935–3943 (2013). Article CAS PubMed PubMed Central Google Scholar * Lorenzen, S. et al. Phase III randomized, double-blind study of paclitaxel with and
without everolimus in patients with advanced gastric or esophagogastric junction carcinoma who have progressed after therapy with a fluoropyrimidine/platinum-containing regimen (RADPAC).
_Int. J. Cancer_, https://doi.org/10.1002/ijc.33025 (2020). * Shen, Y. C. et al. Phase II multicentered study of low-dose everolimus plus cisplatin and weekly 24-hour infusion of high-dose
5-fluorouracil and leucovorin as first-line treatment for patients with advanced gastric cancer. _Oncology_ 87, 104–113 (2014). Article CAS PubMed Google Scholar Download references
ACKNOWLEDGEMENTS The manuscript was edited by Alexandra H. Marshall (Marshall Medical Communications). This work was supported by the National Natural Science Foundation of China (81172487
to L.Liu, and 81500092 to S.Li.), Natural Science Foundation of Shandong Province (ZR201702180008 to L.Liu), and Chinese Postdoctoral Science Foundation (2019M652406 to S.Li.). AUTHOR
INFORMATION Author notes * These authors contributed equally: Yiting Sun, Song Li, Wenbin Yu AUTHORS AND AFFILIATIONS * Department of Medical Oncology, Qilu Hospital, Cheeloo College of
Medicine, Shandong University, Jinan, Shandong, 250012, China Yiting Sun, Song Li, Zeyi Zhao, Jing Gao & Lian Liu * Department of Medical Oncology, National Cancer Center/National
Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, 100021, China Yiting Sun * Department of General
Surgery, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, 250012, China Wenbin Yu, Cheng Chen, Meng Wei & Teng Liu * Animal Laboratory, Qilu Hospital,
Cheeloo College of Medicine, Shandong University, Jinan, Shandong, 250012, China Lanbo Li Authors * Yiting Sun View author publications You can also search for this author inPubMed Google
Scholar * Song Li View author publications You can also search for this author inPubMed Google Scholar * Wenbin Yu View author publications You can also search for this author inPubMed
Google Scholar * Zeyi Zhao View author publications You can also search for this author inPubMed Google Scholar * Jing Gao View author publications You can also search for this author
inPubMed Google Scholar * Cheng Chen View author publications You can also search for this author inPubMed Google Scholar * Meng Wei View author publications You can also search for this
author inPubMed Google Scholar * Teng Liu View author publications You can also search for this author inPubMed Google Scholar * Lanbo Li View author publications You can also search for
this author inPubMed Google Scholar * Lian Liu View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Lian Liu. ETHICS
DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. Edited by N. Barlev SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE LEGENDS SUPPLEMENTARY FIGURE 1 RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sun, Y., Li, S.,
Yu, W. _et al._ _N_6-methyladenosine-dependent pri-miR-17-92 maturation suppresses PTEN/TMEM127 and promotes sensitivity to everolimus in gastric cancer. _Cell Death Dis_ 11, 836 (2020).
https://doi.org/10.1038/s41419-020-03049-w Download citation * Received: 01 June 2020 * Revised: 21 September 2020 * Accepted: 23 September 2020 * Published: 09 October 2020 * DOI:
https://doi.org/10.1038/s41419-020-03049-w SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Euthanasia Counseling | Snopes.comCLAIM: The health care bill currently before Congress mandates that seniors be given euthanasia counseling every five ...
Galaxy fall to rapids, matching longest losing streak in nearly two yearsThe Galaxy’s May slump turned into a full-fledged free fall Sunday when they lost to a team that hadn’t won this season ...
Ole gunnar solskjaer admits anthony martial may be out for the season* RED DEVILS FORWARD MARTIAL SPRAINED HIS KNEE PLAYING FOR FRANCE VS KAZAKHSTAN * FRANCE BOSS DIDIER DESCHAMPS HAD INSIS...
Kneecap announce ‘biggest headline show outside of ireland’ at london arenaTHE CONCERT COMES AFTER THE GROUP REMAINED ON THE LINE-UP FOR GLASTONBURY FESTIVAL SION MORGAN Head of Audience and CASE...
Nh outlook | christa's living legacyNH Outlook Special | 26m 25s The 25th anniversary of the Space Shuttle Challenger. Friday, Jan. 28, 2011 marks the 25th ...
Latests News
N6-methyladenosine-dependent pri-mir-17-92 maturation suppresses pten/tmem127 and promotes sensitivity to everolimus in gastric cancerABSTRACT _N_6-methyladenosine (m6A) is the most common epigenetic RNA modification with essential roles in cancer progre...
Fitness expert shares ‘simple answer’ to losing belly fatWhen looking for exercise and weight-loss inspiration, you might turn to social media and scroll through countless TikTo...
Lucifer season 5 spoilers: will dan find out lucifer’s true identity?Lucifer fans are eager to see the fifth and final season of Lucifer when it lands on Netflix soon. Ahead of the new outi...
Page Not Found很抱歉,你所访问的页面已不存在了。 如有疑问,请电邮[email protected] 你仍然可选择浏览首页或以下栏目内容 : 新闻 生活 娱乐 财经 体育 视频 播客 新报业媒体有限公司版权所有(公司登记号:202120748H)...
Kenya rangers fit tracker to last white giraffe after poachers kill its familyA tiny tracking device has been fitted to a horn of the world’s last known white giraffe in an effort to spare it from p...