Molecular underpinnings of clinical disparity patterns in african american vs. Caucasian american multiple myeloma patients
Molecular underpinnings of clinical disparity patterns in african american vs. Caucasian american multiple myeloma patients"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Caucasian Americans (CA) compared with African Americans (AA) have a twofold increased incidence of multiple myeloma (MM) and have an earlier age of diagnosis. However, there is
sparse information regarding underlying biological differences across racial/ethnic groups. We characterized genetic alterations using a targeted next-generation sequencing assay called
myTYPE, developed at MSKCC, allowing capture of somatic mutations, IgH translocations, gains/losses, and hyperdiploidy. Samples were obtained from the NIH Plasma Cell Dyscrasia Racial
Disparity Cohort. In total, 68 patient samples were successfully sequenced and manually curated based on well-established databases. Of the 68 patient samples (47 CA, 21 AA), 84% had at
least one type of genomic alteration. Importantly, the IgH translocation, t(11;14), was observed more frequently in the AA group (0 vs. 29%, _p_ = 0.001). Known oncogenic somatic
non-synonymous mutations were found in 18 genes and indels in 2 genes. _KRAS_ mutations were the most common mutation found in 16% of patients followed by _NRAS_ and _BRAF_ mutations. _TP53_
somatic mutations appeared to be more common in CA but lacked significance. This proof-of-principle study indicates the presence of varying underlying tumor biology between racial groups
and supports the need of future prospective trials to capture these molecular characteristics. SIMILAR CONTENT BEING VIEWED BY OTHERS COMPREHENSIVE GENETIC ANALYSIS BY TARGETED SEQUENCING
IDENTIFIES RISK FACTORS AND PREDICTS PATIENT OUTCOME IN MANTLE CELL LYMPHOMA: RESULTS FROM THE EU-MCL NETWORK TRIALS Article Open access 16 September 2024 MULTIOMIC PROFILING IDENTIFIES
PREDICTORS OF SURVIVAL IN AFRICAN AMERICAN PATIENTS WITH ACUTE MYELOID LEUKEMIA Article Open access 04 October 2024 GENOMIC ANALYSIS OF PRIMARY PLASMA CELL LEUKEMIA REVEALS COMPLEX
STRUCTURAL ALTERATIONS AND HIGH-RISK MUTATIONAL PATTERNS Article Open access 19 June 2020 INTRODUCTION Despite advancements in the understanding and treatment of multiple myeloma (MM), a
racial disparity in clinical presentation and outcomes remain. Compared with Caucasian Americans (CA), African Americans (AA) matched for socioeconomics, age, and gender have a twofold
increased incidence of MM, have an earlier average age at diagnosis by 5–10 years, and have gained less benefit from the advent of novel agents in the last decade1,2. These differences have
not been shown to be attributable to disparities in access to medical care. In addition, over the past decade, improvements in survival with the introduction of proteasome inhibitors and
immunomodulatory agents is predominantly observed in CA. Costa et al.3 observed improvements in 10-year relative survival rates (RSRs) in all racial groups < 65 years of age and no
improvements for either racial group over 75 years of age. In patients between the ages of 65 and 74 years, CA had an improvement in 10-year RSRs but AA did not. Moreover, although it has
been noted that AA have an increased myeloma-related mortality rate, this is in fact a reflection of the increased incidence of MM in AA rather than worse prognosis. In a pivotal study of
30,000 patients, the authors concluded that AA appear to have a better prognosis compared with CA4. The variation in clinical course suggests an underlying molecular heterogeneity between
races. Despite the increased frequency of MM among AA, most of the known molecular data and association with clinical outcomes, including traditional fluorescence in situ hybridization
(FISH)/cytogenetics and newer NGS methods have been derived from CA cohorts5,6,7,8. At this time there is no single unifying genetic or genomic alteration known to cause MM but there are
multiple alterations frequently identified. Approximately half of MM genomes are hyperdiploid (gain of an additional odd numbered chromosomes)9,10. Most of the non-hyperdiploid MM cases
harbor a translocation involving the immunoglobulin heavy-chain (IgH) gene located on chromosome 149,10. These genetic lesions are thought to be primary events, as they are also found in the
precursor state, monoclonal gammopathy of undetermined significance (MGUS)11. In ~10% of cases, both aberrations co-occur12,13. In general, hyperdiploid MM is associated with an improved
prognosis compared with MM cases with an IgH translocation, except for the cyclin D translocations (t(6;14) and t(11;14)), which are considered neutral14,15. The five most frequent
translocations in descending order are t(11;14), t(4;14), t(14;16), t(14;20), and t(6;14)15,16. Based on karyotyping and interphase FISH, t(4;14), t(14;16), and t(14;20) have been identified
as high-risk primary genetic events, along with the secondary/tertiary events of deletion 17p, deletion 1p32, and 1q gains9,14. The genetic heterogeneity of myeloma is reflected in the
variety of genetic hits including secondary translocations, copy number variants (CNVs), and somatic oncogenic mutations17. To improve our understanding of the underlying biological
mechanisms of the racial disparity in patients with MM, this study used a targeted NGS assay termed myTYPE developed at Memorial Sloan Kettering Cancer Center. myTYPE was specifically
developed to target genomic aberrations known to occur in patients with MM18,19. The myTYPE assay is designed to capture known IgH translocations, hyperdiploidy, CNVs, and somatic mutations
in 120 frequently mutated genes in MM. Using this specific assay we investigated the differences in somatic mutations, translocations, and chromosomal gains/losses between CA and AA MM
patients. METHODS PATIENTS AND TECHINICAL ASSAYS Bone marrow clot sections were obtained from the National Institutes of Health Plasma Cell Dyscrasia Racial Disparity Cohort. A total of 91
pretreatment baseline samples from patients with newly diagnosed MM (NDMM) underwent DNA extraction, 81 samples met DNA quality control (QC) and purity criteria, and underwent NGS library
preparation. Of these, 68 (47 CA, 21 AA) patient samples passed all QC measures for sequencing. In the myTYPE assay, baits were designed to capture the entire IgH locus (where the majority
of the canonical chromosome 14 breakpoints occur) and the partner chromosome, genome-wide single-nucleotide polymorphisms for hyperdiploidy, and other CNVs as well as exons of 120 frequently
mutated genes in MM. With this design, myTYPE detects the IGH translocations and the partner chromosome regardless of which the partner is. In addition, myTYPE detects hyperdiploidy,
arm-level chromosomal gains and losses, as well as the most common and relevant somatic mutations. The target regions from bone marrow clot section samples were amplified and then sequenced
using 126 base-paired end reads using Illumina HiSeq with a mean target depth of 413.5×. Data were analyzed using validated bioinformatic algorithms (Supplementary Methods). Race was
determined from patient self-reporting. Fisher’s exact test was used to calculate two-tailed _p_-values for differences between CA and AA groups. The Bonferroni method was used to correct
for multiple testing and control the family-wise error rate at <0.05. This resulted in a significance threshold of _p_ < 0.0015 for each comparison. RESULTS Of the 68 patient baseline
samples sequenced (47 CA, 21 AA), 57 patients (87%) had at least one genomic alteration (i.e., hyperdiploidy, translocations, chromosomal gains/losses, indels, or somatic non-synonymous
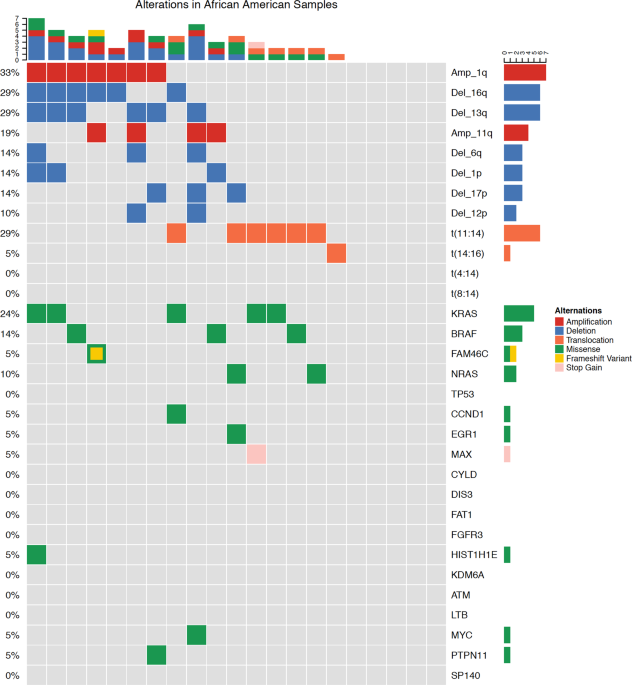
mutations). Of these, 20 (95%) were from AA and 37 (79%) from CA patient samples (Figs. 1 and 2). Putative oncogenic mutations and indels (insertions and deletions) were observed in 19
oncogenic genes (Table 1). _KRAS_ mutations were most common, identified in 16% of patients (13% CA, 24% AA_). NRAS_ (4% CA, 10% AA) and _BRAF_ (2% CA, 14% AA) mutations were the second most
common. _BRAF_ mutations were all V600E non-synonymous mutations and statistically trended to be more common in AA (_p_ = 0.08). _TP53_ somatic mutations also appeared to be more common in
CA (6%) than AA (0%); however, this difference did not reach statistical significance. Oncogenic mutations were also observed in _FAM46C_, _EGR1_, _PTPN11_, _ATM_, _CCND1_, _DIS3_, _FAT1_,
_FGFR3_, _HIST1H1E_, _KDM6A_, _LTB_, _MAX_, _MYC_, and _SP140_. Indels (frameshift and in-frame variants) were identified in _FAM46C_ and _CYLD_. In 11 of the patient samples, no genomic
alterations were observed. Four different translocations involving IgH were identified in patients (Table 2). The t(11;14) occurred significantly more frequently in the AA group (29% vs. 0,
_p_ = 0.0005). This difference remained significant when adjusting for multiple testing. The incidence of t(4;14), t(8;14), and t(14;16) were not significantly different between the CA and
AA groups. Chromosomal gains and losses were identified in many patients and, as expected, approximately half the patients had hyperdiploid myeloma (Table 2). Moreover, del(1p) was observed
more commonly in the AA population (0 vs. 14%, _p_ = 0.037) as well as amp(1q) (13% vs. 33%, _p_ = 0.091); however, neither remained significant when adjusting for multiple testing. There
were no other significant differences between racial groups including patients with hyperdiploid genomes, del(17p), or del(13q). Given the striking observation that t(11;14) was more common
in AA patient samples, IgH translocation data were downloaded from the Multiple Myeloma Research Foundation (MMRF) CoMMpass (Relating Clinical Outcomes in MM to Personal Assessment of
Genetic Profile) Study (research.themmrf.org) to analyze and determine whether racial differences in t(11;14) were observed in a larger cohort of patients20. To the best of our knowledge,
racial differences in translocations in this cohort had not been previously reported. Based on validated racial classification previously reported, 658 samples were evaluable for chromosome
14 translocations (112 AA; 546 CA). No statistically significant differences were observed between races including t(11;14) (Table 3)8. DISCUSSION These results suggest the presence of
genetic heterogeneity between MM racial groups. Our results are consistent with the existing MM molecular literature including the observation that _KRAS_, _NRAS_, _DIS3_, and _TP53_ are
commonly mutated genes in MM8. As described in many malignancies, in MM, _TP53_ mutations indicate a poor prognosis and shorter survival; however, the effects of other mutations are not well
characterized21,22. We observed _TP53_ mutations more frequently in CA, which is consistent with the findings from a recent study that also observed significantly higher _TP53_ mutation
rates among CA MM cases8. Located on chromosome 17p13.1, _TP53_ encodes for p53 tumor suppressor protein mediating multiple cell cycle pathways including apoptosis, cell cycle arrest, and
inhibition of angiogenesis23. In MM, _TP53_ mutations are a rare occurrence at diagnosis; however, the incidence increases as patients are treated. It is often associated with poor prognosis
and accounts for a significantly lower survival rate24. This finding suggests potentially one etiology for the worse prognosis observed in CA4. Furthermore, this may have important clinical
implications in terms of targeted drug development. For example, compounds in various stages of development including MDM2 inhibitors, focus on restoring wild-type p53 activity25. A few of
these agents have proceeded to first-in-human phase 1 interventional MM clinical trials26. Importantly, in our cohort, t(11;14) was found to be significantly more frequent in the AA group
(_p_ = 0.0005)_._ t(11;14) is a frequent translocation in MM found in about 15–20% of patients10,27,28. Recently, the Mayo Clinic group using traditional FISH methods observed a similar
association with t(11;14) in their AA cohort29. They evaluated 881 patients with monoclonal gammopathies and found that the probability of having t(11;14) (or t(14;16)/t(14;20) was
significantly higher in the 120 patients with highest AA ancestry (≥80%) compared with individuals with lowest levels of AA ancestry. This finding helps to confirm that our results are not
random, and that NGS methods can be used to confirm traditional FISH findings. The t(11;14)(q13;q32) results in upregulation of cyclin D1, thus promoting cell cycle progression10. Most data
support that the presence of t(11;14) is associated with neutral or standard prognostic risk, and that it may confer improved survival and response to treatment compared with the other
commonly observed IgH translocations10,28,30,31. However, a few smaller studies suggest that patients with MM harboring t(11;14) may not have the same prognosis as patients with other
standard risk features32. Interestingly, recent work suggests that AA patients with t(11;14) showed a trend toward shorter median progression-free survival (PFS) compared with AA without the
presence of t(11;14); however, t(11;14) did not impact PFS in non-AA patients33. More importantly, this genetic alteration also has significant implications for drug development. More
recently, it has been observed that patients with this translocation are much more likely to respond to BLC-2 inhibitors, and that this genotype is associated with increased expression of
the anti-apoptotic protein BCL-2 compared with pro-apoptotic family members. For example, the BCL-2 inhibitor, venetoclax, as monotherapy is associated with a response rate of 40% in
patients with t(11;14) compared with 21% in all comers34. In short, we were unable to confirm our finding in the CoMMpass cohort; however, the confirmatory finding in the Mayo AA cohort
reinforces this finding, which has important implications in the AA population and precision drug development. Deletion of chromosome 1p, (del(1p)), which is associated with a poor
prognosis, in our cohort, appeared to also be more frequent (_p_ = 0.037) along with amp(1q) in AA; however, these were not significant after adjusting for multiple testing and no
differences were observed in the CoMMpass cohort8,35. Although t(11;14) is thought to be a primary event as it is observed in the early precursor state of MGUS, del(1p) is thought to be a
secondary event further driving MM clonal evolution36. Therefore, this finding may be biased by the timing of when patients were diagnosed. In addition to risk prognostication, the
differences in somatic mutations among races may have significant implications in the development of targeted therapies. For example, the _BRAF__V600E_ mutation is a frequent and
well-described driver mutation in melanoma and hairy cell leukemia with approximate response rates of 50–60% and 96–100%, respectively, to _BRAF_ inhibition with tyrosine kinase
inhibitors37,38,39. Interestingly, _BRAF_ mutations occur in ~5% of MM cases with tumors that respond to tyrosine kinase inhibition40,41. Albeit a rare driver mutation in MM, it is an
important druggable target. Interestingly, we observed a higher rate of _BRAF__V600E_ mutations in AA (14%) compared with CA (2%); however, this finding was not statistically significant and
was not observed in the independent CoMMpass cohort. Our findings along with other works suggest that the incidence of the various prognostic primary genetic events is not significantly
different between races (e.g., t(4;14) and hyperdiploidy). Rather, the differences between races are predominantly events known to occur later in disease evolution. The molecular pathology
of MM changes overtime and multiple clonal competitions occur in the cancer cell population through branching evolution36. Clonal and sub-clonal evolution occurs in the context of pressures
present in the tumor micro-environment including treatment effect creating a branching nonlinear pathway of multiclonal MM development5. Based on this, one might speculate that the primary
pathogenic events are similar across races, whereas the ensuing disease evolution follows slightly different trajectories, shaped by inter-racial differences in tumor–host interactions42.
This study is limited by the small number of patients. However, it expands upon the limited molecular data from AA with MM. We plan to further study and expand on these findings by examining
the genetic alterations present and associated clinical outcome differences between races in patients with smoldering MM and comparison with NDMM. Our current actively enrolling study
Carfilzomib, Lenalidomide, and Dexamethasone in High Risk Smoldering Multiple Myeloma will be the vehicle to aid in answering these important questions and thus far has shown impressive
results at interim analysis (https://clinicaltrials.gov/ct2/show/NCT01572480)43,44. This information will add to our knowledge of the clonal evolution of MM, prognostic value of genetic
data, and elucidate potential differences in smoldering myeloma compared with NDMM in terms of race. CONCLUSIONS The findings of this work significantly contribute to the understanding of
molecular differences between races in MM, in a relative knowledge desert. These findings argue for more enrichment of AA patients in prospective MM treatment trials and characterization of
molecular profiles. REFERENCES * Waxman, A. J. et al. Racial disparities in incidence and outcome in multiplemyeloma. _a population-based study. Blood_ 116, 5501–5506
https://doi.org/10.1182/blood-2010-07-298760 (2010). Article CAS PubMed Google Scholar * Greenberg, A. J. & Rajkumar, S. V. Elucidating disparities across racial and ethnic groups in
multiple myeloma patients. _Int. J. Hematol._ 95, 453–454, https://doi.org/10.1007/s12185-012-1040-y (2012). Article PubMed Google Scholar * Costa, L. J. et al. Recent trends in multiple
myeloma incidence and survival by age, race, and ethnicity in the United States. _Blood Adv._ 1, 282–287, https://doi.org/10.1182/bloodadvances.2016002493 (2017). Article PubMed PubMed
Central Google Scholar * Landgren, O. et al. African-American multiple myeloma patients have a better survival than Caucasian patients: a population-based study including 28,636 patients.
_Blood_ 114, 1832 (2009). Google Scholar * Bolli, N. et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. _Nat. Commun._ 5, 2997,
https://doi.org/10.1038/ncomms3997 (2014). Article CAS PubMed Google Scholar * Chapman, M. A. et al.Initial genome sequencing and analysis of multiple myeloma. _Nature_ 471, 467–472,
https://doi.org/10.1038/nature09837 (2011). Article CAS PubMed PubMed Central Google Scholar * Lohr, J. G. et al.Widespread genetic heterogeneity in multiple myeloma: implications for
targeted therapy. _Cancer Cell._ 25, 91–101, https://doi.org/10.1016/j.ccr.2013.12.015 (2014). Article CAS PubMed PubMed Central Google Scholar * Manojlovic, Z. et al.Comprehensive
molecular profiling of 718 multiple myelomas reveals significant differences in mutation frequencies between African and European descent cases. _PLoS Genet._ 13, e1007087,
https://doi.org/10.1371/journal.pgen.1007087 (2017). Article CAS PubMed PubMed Central Google Scholar * Pawlyn, C. & Morgan, G. J. Evolutionary biology of high-risk multiple
myeloma. _Nat. Rev. Cancer_ 17, 543–556, https://doi.org/10.1038/nrc.2017.63 (2017). Article CAS PubMed Google Scholar * Fonseca, R. et al. Myeloma and the t(11;14)(q13; q32); evidence
for a biologically defined unique subset of patients. _Blood_ 99, 3735–3741 (2002). Article CAS PubMed Google Scholar * Bergsagel, P. L. & Kuehl, W. M. Molecular pathogenesis and a
consequentclassification of multiple myeloma. _J. Clin. Oncol_ 23, 6333–6338, https://doi.org/10.1200/jco.2005.05.021 (2005). Article CAS PubMed Google Scholar * Smadja, N. Vr, Bastard,
C., Brigaudeau, C., Leroux, D. & Fruchart, C. Hypodiploidy is a major prognostic factor in multiple myeloma. _Blood_ 98, 2229–2238 (2001). Article CAS PubMed Google Scholar *
Kazandjian, D., Mailankody, S., Korde, N. & Landgren, O. Smoldering multiple myeloma: pathophysiologic insights, novel diagnostics, clinical risk models, and treatment strategies. _Clin.
Adv. Hematol. Oncol._ 12, 578–587 (2014). PubMed Google Scholar * Robiou du Pont, S. et al. Genomics of multiple myeloma. _J. Clin. Oncol._ 35, 963–967,
https://doi.org/10.1200/JCO.2016.70.6705 (2017). Article PubMed Google Scholar * Morgan, G. J., Walker, B. A. & Davies, F. E. The genetic architecture of multiple myeloma. _Nat. Rev.
Cancer_ 12, 335–348 (2012). Article CAS PubMed Google Scholar * Walker, B. A. et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. _Blood_
132, 587–597 https://doi.org/10.1182/blood-2018-03-840132 (2018). * Kazandjian, D. Multiple myeloma epidemiology and survival: a unique malignancy. _Semin. Oncol._ 43, 676–681,
https://doi.org/10.1053/j.seminoncol.2016.11.004 (2016). * Hill, E. M. et al. Molecular underpinnings of clinical disparity patterns in AfricanAmerican (AA) versus Caucasian American (CA)
multiple myeloma (MM)patients. _J. Clin. Oncol_ 36, 8036, https://doi.org/10.1200/JCO.2018.36.15_suppl.8036 (2018). Article Google Scholar * Malin Hultcrantz, V. Y., et al. myTYPE targeted
next generation sequencing assay for detection of IgH translocations and copy number alterations in multiple myeloma
https://learningcenter.ehaweb.org/eha/2018/stockholm/214976/malin.hultcrantz.mytype.targeted.next.generation.sequencing.assay.for.html (Abstract PF525 presented at European Hematology
Association Annual Meeting, Stockholm. EHA Learning Center. 2018; 214976). * Lonial, S. et al.Interim analysis of the Mmrf Commpass Trial: identification of novel rearrangements potentially
associated with disease initiation and progression. _Blood_ 124, 722–722 (2014). Google Scholar * Alexandrov, L. B. et al.Signatures of mutational processes in human cancer. _Nature_ 500,
415–421, https://doi.org/10.1038/nature12477 (2013). Article CAS PubMed PubMed Central Google Scholar * Walker, B. A. et al. Mutational spectrum, copy number changes, and outcome:
results of a sequencing study of patients with newly diagnosed myeloma. _J. Clin. Oncol._ 33, 3911–3920, https://doi.org/10.1200/jco.2014.59.1503 (2015). Article CAS PubMed Google Scholar
* Vousden, K. H. & Lu, X. Live or let die: the cell’s response to p53. _Nat. Rev. Cancer_ 2, 594–604, https://doi.org/10.1038/nrc864 (2002). Article CAS PubMed Google Scholar *
Chng, W. J. et al.Clinical significance of TP53 mutation in myeloma. _Leukemia_ 21, 582–584, https://doi.org/10.1038/sj.leu.2404524 (2007). Article CAS PubMed Google Scholar * Zhao, D.,
Tahaney, W. M., Mazumdar, A., Savage, M. I. & Brown, P. H. Molecularly targeted therapies for p53-mutant cancers. _Cell. Mol. Life Sci._ 74, 4171–4187,
https://doi.org/10.1007/s00018-017-2575-0 (2017). Article CAS PubMed PubMed Central Google Scholar * Tisato, V., Voltan, R., Gonelli, A., Secchiero, P. & Zauli, G. MDM2/X inhibitors
under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. _J. Hematol. Oncol._ 10, 133, https://doi.org/10.1186/s13045-017-0500-5
(2017). Article CAS PubMed PubMed Central Google Scholar * Sawyer, J. R.The prognostic significance of cytogenetics and molecular profiling in multiple myeloma. _Cancer Genet._ 204,
3–12, https://doi.org/10.1016/j.cancergencyto.2010.11.002 (2011). Article PubMed Google Scholar * Avet-Loiseau, H. et al.Genetic abnormalities and survival in multiple myeloma: the
experience of the Intergroupe Francophone du Myelome. _Blood_ 109, 3489–3495, https://doi.org/10.1182/blood-2006-08-040410 (2007). Article CAS PubMed Google Scholar * Baughn, L. B. et
al.Differences in genomic abnormalities among African individuals with monoclonal gammopathies using calculated ancestry. _Blood Cancer J._ 8, 96, https://doi.org/10.1038/s41408-018-0132-1
(2018). Article PubMed PubMed Central Google Scholar * Moreau, P. et al. Recurrent 14q32 translocations determine the prognosis of multiple myeloma, especially in patients receiving
intensive chemotherapy. _Blood_ 100, 1579–1583, https://doi.org/10.1182/blood-2002-03-0749 (2002). Article CAS PubMed Google Scholar * Leiba, M. et al. Translocation t(11;14) in newly
diagnosed patients with multiple myeloma: Is it always favorable?. _Genes Chromosomes Cancer_ 55, 710–718, https://doi.org/10.1002/gcc.22372 (2016). Article CAS PubMed Google Scholar *
Lakshman, A. et al.Natural history of t(11;14) multiple myeloma. _Leukemia_ 32, 131–138, https://doi.org/10.1038/leu.2017.204 (2018). Article CAS PubMed Google Scholar * Gasparetto, C.
J. et al.Differential effect of t(11;14) abnormality on survival and depth of response in African American (AA) and non-AA (NAA) patients (Pts) with newly diagnosed multiple myeloma (NDMM)
in the Connect® MM Registry. _Blood_ 130, 3101–3101 (2017). Google Scholar * Kortüm, K. M. & Einsele, H. First targeted therapy in multiple myeloma. _Blood_ 130, 2359–2360,
https://doi.org/10.1182/blood-2017-09-805341 (2017). Article CAS PubMed Google Scholar * Ouyang, J., Gou, X., Ma, Y., Huang, Q. & Jiang, T.Prognostic value of 1p deletion for
multiple myeloma: a meta-analysis. _Int. J. Lab. Hematol._ 36, 555–565, https://doi.org/10.1111/ijlh.12189 (2014). Article CAS PubMed Google Scholar * Manier, S. et al.Genomic complexity
of multiple myeloma and its clinical implications. _Nat. Rev. Clin. Oncol._ 14, 100–113, https://doi.org/10.1038/nrclinonc.2016.122 (2017). Article CAS PubMed Google Scholar * Chapman,
P. B. et al.Improved survival with Vemurafenib in melanoma with BRAF V600E mutation. _New Engl. J. Med._ 364, 2507–2516, https://doi.org/10.1056/NEJMoa1103782 (2011). Article CAS PubMed
Google Scholar * Hauschild, A. et al.Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. _Lancet_ 380, 358–365,
https://doi.org/10.1016/s0140-6736(12)60868-x (2012). Article CAS PubMed Google Scholar * Tiacci, E. et al.Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. _New Engl.
J. Med._ 373, 1733–1747, https://doi.org/10.1056/NEJMoa1506583 (2015). Article CAS PubMed Google Scholar * Andrulis, M. et al.Targeting the BRAF V600E mutation in multiple myeloma.
_Cancer Discov._ 3, 862–869, https://doi.org/10.1158/2159-8290.Cd-13-0014 (2013). Article CAS PubMed Google Scholar * Raje, N. et al.Vemurafenib (VEM) in relapsed refractory multiple
myeloma harboring BRAFV600 mutations (V600m): a cohort of the Histology-Independent VE-Basket Study. _Blood_ 126, 4263–4263 (2015). Google Scholar * Özdemir, B. C. & Dotto, G.-P. Racial
differences in cancer susceptibility andsurvival: more than the color of the skin?. _Trends Cancer_ 3, 181–197, https://doi.org/10.1016/j.trecan.2017.02.002 (2017). Article PubMed PubMed
Central Google Scholar * Kazandjian, D. et al. Sustained minimal residual disease negativity in newly diagnosed multiple myeloma (NDMM) patients treated with Carfilzomib (CFZ),
Lenalidomide (LEN), and Dexamethasone (DEX) followed by 2 years of lenalidomide maintenance (CRd-R): updated results of a phase 2 study. _Blood_ 128, 4527–4527 (2016). Google Scholar *
Mailankody, S. et al. Baseline mutational patterns and sustained MRD negativityin patients with high-risk smoldering myeloma. _Blood Adv_ 1, 1911–1918,
https://doi.org/10.1182/bloodadvances.2017005934 (2017). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank Dr. Martin Mendoza and the Office
of Minority Affairs, U.S. Food and Drug Administration and the Memorial Sloan Kettering Core Grant (P30 CA008748) for grant support in conducting this work. The views expressed in this
manuscript are those of the author and do not reflect the official policy of the Department of Army/Navy/Air Force, Department of Defense, or U.S. Government. AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Myeloma Program, Lymphoid Malignancies Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA Dickran Kazandjian &
Elizabeth Hill * Myeloma Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York City, NY, USA Malin Hultcrantz, Evan H. Rustad, Theresia Akhlaghi, Neha Korde,
Sham Mailankody & Ola Landgren * Epidemiology and Biostatistics, Memorial Sloan Kettering Cancer Center, New York City, NY, USA Venkata Yellapantula & Elli Papaemmanuil * Department
of Hematology-Oncology, Walter Reed National Military Medical Center, Bethesda, MD, USA Alex Dew & Mary Kwok * Hematology Section, Department of Laboratory Medicine Clinical Center,
National Institutes of Health, Bethesda, MD, USA Irina Maric Authors * Dickran Kazandjian View author publications You can also search for this author inPubMed Google Scholar * Elizabeth
Hill View author publications You can also search for this author inPubMed Google Scholar * Malin Hultcrantz View author publications You can also search for this author inPubMed Google
Scholar * Evan H. Rustad View author publications You can also search for this author inPubMed Google Scholar * Venkata Yellapantula View author publications You can also search for this
author inPubMed Google Scholar * Theresia Akhlaghi View author publications You can also search for this author inPubMed Google Scholar * Neha Korde View author publications You can also
search for this author inPubMed Google Scholar * Sham Mailankody View author publications You can also search for this author inPubMed Google Scholar * Alex Dew View author publications You
can also search for this author inPubMed Google Scholar * Elli Papaemmanuil View author publications You can also search for this author inPubMed Google Scholar * Irina Maric View author
publications You can also search for this author inPubMed Google Scholar * Mary Kwok View author publications You can also search for this author inPubMed Google Scholar * Ola Landgren View
author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHORS Correspondence to Dickran Kazandjian or Ola Landgren. ETHICS DECLARATIONS CONFLICT OF
INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL_METHODS-REVISED-CLEAN RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative
Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the
original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in
the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended
use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kazandjian, D., Hill, E., Hultcrantz, M. _et al._ Molecular underpinnings of
clinical disparity patterns in African American vs. Caucasian American multiple myeloma patients. _Blood Cancer Journal_ 9, 15 (2019). https://doi.org/10.1038/s41408-019-0177-9 Download
citation * Received: 29 September 2018 * Revised: 18 December 2018 * Accepted: 14 January 2019 * Published: 04 February 2019 * DOI: https://doi.org/10.1038/s41408-019-0177-9 SHARE THIS
ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard
Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Trump administration plans to require immigrants to registerNewsletters ePaper Sign in HomeIndiaKarnatakaOpinionWorldBusinessSportsVideoEntertainmentDH SpecialsOperation SindoorNew...
Imexpg502000 - approval: specimen letter - hmrc internal manualIMEXPG502000 - APPROVAL: SPECIMEN LETTER Dear Sirs APPROVAL OF PIPELINE FROM [ FIELD] TO [ TERMINAL] I am directed by th...
The State We're In | PBSSHARE THIS SHOW * Link Copied to Clipboard HOW TO WATCH THE STATE WE'RE IN The State We're In is available to ...
Edited image of greta thunberg shared amid ‘toolkit’ controversyA reverse image search led us to the original image, which was tweeted by Thunberg on 22 January 2019, identifying the l...
Trump's mideast tour: over $2 trillion in business deals, markets gain momentumMore than $2 trillion in business deals “were locked in” during the historic four-day official to three GCC countries by...
Latests News
Molecular underpinnings of clinical disparity patterns in african american vs. Caucasian american multiple myeloma patientsABSTRACT Caucasian Americans (CA) compared with African Americans (AA) have a twofold increased incidence of multiple my...
Joe sugg and dianne buswell: debbie mcgee addresses their romanceJoe Sugg, 27 and Dianne Buswell, 29, were partnered together on the hit BBC show Strictly Come Dancing 2018 and now form...
Battle of Britain hero who led charmed life dies aged 98It was August 16, 1940, and the civilian population that lived along the coast by The Solent had a grandstand view of on...
Channelnews : acer rocks into ces 2023 with new macbook air competitorAcer has rocked into CES 2023, with a whole new line-up of business PC’s Predator gaming machines featuring Intel’s 13th...
A computational program for automated surgical planning of fenestrated endovascular repairABSTRACT An Abdominal Aortic Aneurysm (AAA) is a dilation of the aorta at the level of the abdomen. To reduce the risk o...