Somatic and germline mutations in the tumor suppressor gene park2 impair pink1/parkin-mediated mitophagy in lung cancer cells
Somatic and germline mutations in the tumor suppressor gene park2 impair pink1/parkin-mediated mitophagy in lung cancer cells"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT _PARK2_, which encodes Parkin, is a disease-causing gene for both neurodegenerative disorders and cancer. Parkin can function as a neuroprotector that plays a crucial role in the
regulation of mitophagy, and germline mutations in _PARK2_ are associated with Parkinson’s disease (PD). Intriguingly, recent studies suggest that Parkin can also function as a tumor
suppressor and that somatic and germline mutations in _PARK2_ are associated with various human cancers, including lung cancer. However, it is presently unknown how the tumor suppressor
activity of Parkin is affected by these mutations and whether it is associated with mitophagy. Herein, we show that wild-type (WT) Parkin can rapidly translocate onto mitochondria following
mitochondrial damage and that Parkin promotes mitophagic clearance of mitochondria in lung cancer cells. However, lung cancer-linked mutations inhibit the mitochondrial translocation and
ubiquitin-associated activity of Parkin. Among all lung cancer-linked mutants that we tested, A46T Parkin failed to translocate onto mitochondria and could not recruit downstream mitophagic
regulators, including optineurin (OPTN) and TFEB, whereas N254S and R275W Parkin displayed slower mitochondrial translocation than WT Parkin. Moreover, we found that deferiprone (DFP), an
iron chelator that can induce mitophagy, greatly increased the death of A46T Parkin-expressing lung cancer cells. Taken together, our results reveal a novel mitophagic mechanism in lung
cancer, suggesting that lung cancer-linked mutations in _PARK_2 are associated with impaired mitophagy and identifying DFP as a novel therapeutic agent for _PARK2_-linked lung cancer and
possibly other types of cancers driven by mitophagic dysregulation. You have full access to this article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS PARKIN
INHIBITS PROLIFERATION AND MIGRATION OF BLADDER CANCER VIA UBIQUITINATING CATALASE Article Open access 29 February 2024 KNOCKING OUT ALPHA-SYNUCLEIN IN MELANOMA CELLS DYSREGULATES CELLULAR
IRON METABOLISM AND SUPPRESSES TUMOR GROWTH Article Open access 04 March 2021 KNOCKING OUT ALPHA-SYNUCLEIN IN MELANOMA CELLS DOWNREGULATES L1CAM AND DECREASES MOTILITY Article Open access 07
June 2023 INTRODUCTION Parkin is an important E3 ubiquitin ligase that is activated by phosphorylated ubiquitin and promotes the polyubiquitination of various substrates [1]. Under normal
conditions, Parkin localizes throughout the nucleus and cytosol, and it can translocate onto damaged mitochondria to promote the ubiquitination of mitochondrial proteins and trigger
mitophagy under stress conditions [2]. Loss-of-function mutations in _PARK2_, the gene encoding Parkin, cause Parkinson’s disease (PD) [3]. However, Parkin knockout mice did not display any
neurodegenerative phenotype but developed spontaneous hepatocellular carcinoma and tumor growth [4, 5], indicating the complexity of Parkin’s functions and associated disease mechanisms.
Moreover, the expression level of Parkin was found to be decreased in several human cancers, including lung cancer [6, 7], and somatic mutations in _PARK2_ were found in glioblastoma and
lung cancer [8, 9]. Importantly, overexpression of Parkin inhibited cell growth and proliferation in multiple cancer cell lines [10,11,12], indicating that Parkin can act as a tumor
suppressor and that its dysfunction may contribute to oncogenesis in somatic cells. Autophagy is an important degradation pathway responsible for cell homeostasis [13]. Although the role of
autophagy in cancer cell survival and death has been extensively explored, the relationship between cancer and the selective forms of autophagy, including mitophagy, is not yet well
understood [14]. Parkin acts as a crucial mediator in mitophagy regulation [2, 14]. Recent research advances in mitophagy have revealed that under normal conditions, Parkin displays an
autoinhibited state, since the active site of Parkin is sealed by the REP domain [15], whereas under disease conditions or mitochondrial stress, Parkin is phosphorylated by mitochondrial
PINK1 at the N-terminal ubiquitin-like (UBL) domain and interacts with phosphorylated ubiquitin on the mitochondrial outer membrane [16]. After a series of conformational changes, the RING
finger E3 catalytic center is exposed, and Parkin acts as an activated E3 ubiquitin ligase [2, 15]. According to recent studies, Parkin can facilitate the ubiquitination of outer
mitochondrial membrane (OMM) proteins and therefore enhance the recognition of polyubiquitinated mitochondria by mitophagic receptors, such as optineurin (OPTN) or NDP52 [17, 18]. These
receptors can bind to autophagosomal proteins, such as LC3 and target ubiquitinated mitochondria for lysosomal degradation [19]. Moreover, a recent report showed that TFEB, the master
transcription factor regulating the expression of autophagic and lysosomal genes, is a downstream regulator of mitophagy [20]. The above process, which is very powerful for clearing damaged
mitochondrial and is the most well-understood form of mitophagy, is called PINK1/Parkin-mediated mitophagy [2]. According to previous studies, somatic _PARK2_ mutations in lung cancer
include the A46T, N254S and H279P mutations [8], while germline mutations in _PARK2_ are found in familial lung cancer [9]. It is worth noting that all these lung cancer-linked mutations are
highly enriched in ubiquitin-associated domains of Parkin. In detail, the A46T mutation occurs in the UBL domain, whereas the N254S, R275W and H279P mutations occur in the RING1 domain of
Parkin [8, 9]. These findings suggest that Parkin-associated ubiquitin regulation may contribute to the disease pathogenesis underlying lung cancer. However, whether these mutations affect
Parkin-mediated mitophagy and the potential role of this alteration in lung cancer tumorigenesis remain unknown. In this study, we showed that these lung cancer-linked mutations abolish the
recognition of damaged mitochondria by autophagosomes due to the loss of mitochondrial ubiquitination driven by Parkin. Importantly, we identified that deferiprone (DFP), which is known as a
mitophagy enhancer [21], can induce cell death in lung cancer cells expressing mutant Parkin. MATERIALS AND METHODS PLASMID CONSTRUCTION AND SIRNAS The FLAG, FLAG-Parkin, HA-Ub, GFP-OPTN,
mCherry-C1 and GFP-TFEB plasmids were described before [22,23,24,25,26,27]. mCherry-Parkin was generously provided by Richard Youle (Addgene #23956); BFP-mito was generously provided by Gia
Voeltz (Addgene #49151). FLAG-tagged and mCherry-tagged A46T, N254S and R275W Parkin mutants were generated by site-directed mutagenesis with a MutanBEST kit (Takara, Shiga, Japan).
pcDNA3.1-mt-Keima was generated by inserting mKeima cDNA into the vector at the _Kpn_I and _EcoR_I sites. All constructs were verified by sequencing. Small interfering RNAs targeting human
OPTN and PINK1 were synthesized by Shanghai GenePharma. CELL CULTURE AND TREATMENTS Human alveolar epithelial cells (A549) were cultured in DMEM (Gibco, Grand Island, NY, USA) containing 10%
FBS (Gibco), 100 U/mL penicillin and 100 mg/mL streptomycin (Gibco). Cells were transfected with DNA plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) 24 h after splitting
and with siRNA using Lipofectamine RNAiMAX (Invitrogen) upon splitting. Propidium iodide (PI, Sigma, St. Louis, MO, USA; 0.5 mg/mL) staining was used to detect dead cells. Antimycin A1,
chloroquine (CQ), CCCP and Hoechst were purchased from Sigma. Oligomycin was purchased from Calbiochem. Bafilomycin A1 (Bafi A1) was obtained from Selleckchem, and MitoTracker Red CMXRos was
purchased from Thermo Fisher Scientific (Waltham, MA, USA). IMMUNOBLOT ANALYSIS AND IMMUNOFLUORESCENCE For immunoblot analysis, cells were lysed with cell lysis buffer [22, 25, 27].
Proteins were then separated by SDS–PAGE and transferred onto PVDF membranes (Millipore, Billerica, MA, USA). The primary antibodies anti-GAPDH and anti-Hsp60 were purchased from
Proteintech. Horseradish peroxidase-conjugated sheep anti-mouse antibody (Jackson ImmunoResearch Laboratories, PA, USA) was used as the secondary antibody. Proteins were detected using an
ECL detection kit (Thermo Fisher Scientific). Immunoblot analysis was performed with a standard protocol as previously reported [28,29,30,31,32,33]. For immunofluorescence, cells were fixed
with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked with 0.1% FBS. Then, cells were incubated sequentially with primary antibodies and secondary antibodies. Anti-HA
(Santa Cruz, CA, USA), anti-FLAG (Sigma) and anti-TOM20 (Proteintech, Rosemont, IL, USA) antibodies were used as primary antibodies. Alexa Fluor 405-conjugated goat anti-rabbit IgG (H+L)
(Invitrogen) and Alexa Fluor 594-conjugated goat anti-mouse IgG (Proteintech) were used as secondary antibodies. Eventually, cells were visualized using a Nikon [34] or Zeiss LSM710 confocal
microscope [35, 36]. Briefly, the imaging analysis was performed by researchers who were blinded to the study, and fluorescence was quantified using ImageJ software. For live cell imaging,
cells were stained with MitoTracker Red CMXRos and Hoechst as described in Fig. S4. STATISTICAL ANALYSIS Parkin and OPTN mitochondrial localization was determined by visual inspection in 10
min intervals. PI-positive cells were considered nonviable. Data are presented as the means ± SDs. Comparisons were performed using Prism 6.0 (GraphPad Software). _P_ < 0.05 was
considered statistically significant. RESULTS BOTH SOMATIC AND GERMLINE MUTATIONS OF PARKIN IN LUNG CANCER IMPAIR PARKIN-MEDIATED MITOPHAGY Parkin, the E3 ubiquitin ligase, has been
identified as a tumor suppressor in several cancers, including lung cancer. We were interested in several _PARK2_ lung cancer mutations, including the somatic mutations A46T and N254S, and a
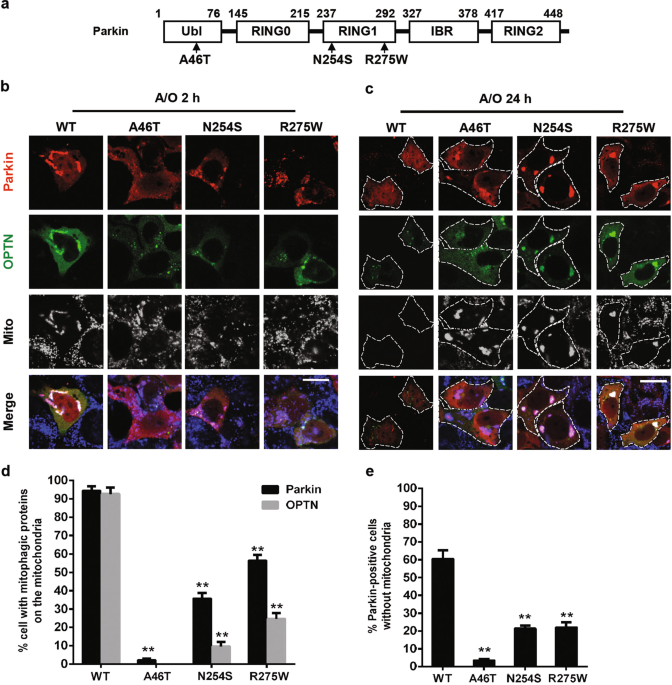
recurrent germline mutation, R275W (Fig. 1a) [8, 9]. To elucidate the effect of these Parkin mutations on mitophagy, we investigated the mitochondrial translocation of Parkin and
mitochondrial clearance in mutant Parkin-overexpressing lung cancer (A549) cells. As shown in Fig. 1, wild-type (WT) Parkin translocated onto damaged mitochondria, followed by mitochondrial
recruitment of the mitophagy receptor OPTN following A/O treatment, a standard method to induce cellular mitophagy [17]. However, Parkin with the lung cancer-related mutations—A46T, N254S
and R275W—showed delayed mitochondrial translocation to different degrees (Fig. 1b, d). Long-term mitophagy induction resulted in complete clearance of mitochondria in cancer cells with WT
Parkin. In contrast, mutant Parkin-expressing cells showed impaired mitochondrial clearance (Fig. 1c, e). PINK1/PARKIN-MEDIATED MITOPHAGY IS BLOCKED BY PARKIN MUTATIONS IN LUNG CANCER CELLS
Although PINK1/Parkin-mediated mitophagy has been extensively investigated, the relationship between mitophagy and lung tumorigenesis remains elusive. To understand the role of
PINK1/Parkin-mediated mitophagy in lung cancer cells, we used a long-term mitophagic degradation assay and investigated the turnover of mitochondrial proteins. Immunoblot analysis showed
that the level of the mitochondrial matrix protein Hsp60 was decreased dramatically in cells expressing WT Parkin (Fig. 2a). Depletion of the mitophagic receptor OPTN caused a reduction in
mitochondrial degradation, even in the presence of WT Parkin, which suggested that OPTN is a downstream mitophagic regulator of Parkin in lung cancer cells. Consistent with the fluorescence
results shown in Fig. 1, A46T and N254S mutant Parkin had no significant effect on mitochondrial degradation, regardless of whether OPTN was knocked down (Figs. 2a and S1a). To verify the
effect of PINK1 on Parkin-associated mitophagy in lung cancer cells, we knocked down the crucial mitochondrial initiator PINK1, which is reported to phosphorylate Parkin and ubiquitin on
mitochondria [2]. Compared to cells with endogenous PINK1 expression, PINK1-deficient lung cancer cells did not exhibit mitochondrial clearance upon A/O or CCCP treatment (Figs. 2b and S1b).
To further test whether the degradation of the mitochondrial protein Hsp60 occurs through autophagy but not other degradation systems, we blocked the autophagy pathway with Bafi A1 or CQ.
The results showed that these autophagy inhibitors abolished Parkin-mediated mitophagy (Fig. 2c). LUNG CANCER-LINKED MUTATIONS ABOLISH MITOCHONDRIAL RECRUITMENT OF PARKIN AND UBIQUITIN AND
DIMINISH NUCLEAR TRANSLOCATION OF TFEB DURING MITOPHAGY To further gain mechanistic insights into the failure of mitophagy driven by these Parkin mutations, we next performed a time-course
experiment to detect the speed of translocation of Parkin and OPTN onto mitochondria during mitophagy. As shown in Fig. 3, WT Parkin rapidly translocated to mitochondria in ~2 h, while N254S
Parkin finished this process at 3 h. Strikingly, A46T Parkin did not show strong mitochondrial localization within similar times (Fig. 3a, b). Therefore, we conclude that lung cancer-linked
UBL mutations in Parkin lead to a significant failure of mitochondrial Parkin and receptor recruitment, whereas RING 1 mutations delay recruitment. We next examined the translocation of
TFEB, which reflects TFEB activity, during Parkin-mediated mitophagy, since it has been shown that TFEB functions downstream of Parkin during mitophagy [20]. We found that translocation of
Parkin onto mitochondria was accompanied by TFEB nuclear translocation (Fig. 4a). However, the lung cancer-related Parkin mutations showed different effects. TFEB was expressed completely in
the cytoplasm in cells with A46T Parkin expression and partially in the nucleus in cells with N254S Parkin expression (Fig. 4a). As an E3 ubiquitin ligase, Parkin is known to ubiquitinate
damaged mitochondria and promote their clearance via autophagy. To determine the ubiquitin ligase activity of mutant Parkin, we tested the ubiquitination of mitochondria. The lung
cancer-related Parkin mutants failed to ubiquitinate damaged mitochondria as WT Parkin did (Figs. 4b and S2). Our observation indicates that mutation of Parkin impaired its E3 ligase
function in mitochondria. LUNG CANCER MUTANT PARKIN ATTENUATES MITOPHAGIC FLUX Based on the observation that mutated Parkin exhibits greatly delayed mitochondrial translocation and defective
clearance of Hsp60, we next visualized mitophagic flux using a fluorescence-sensitive probe, mt-Keima. According to previous studies, mt-Keima is a mitochondria-located fluorescent protein
that is pH-stable. Keima emits different signals with changes in pH. The signal peaks at 586 nm in acidic conditions, such as in the lysosomal lumen, or at 440 nm in neutral pH conditions.
Therefore, acidic autolysosomes containing mitochondria, which reflects mitophagy, can be detected as “561 nm” signals. Meanwhile, nonlysosomal mitochondria, which reflect the pool of
mitochondria that are not targeted by mitophagy, can be detected as “458 nm” signals [37, 38]. We found that 561 nm signals existed when mitochondria were damaged for a short time (Fig. 5,
left region, and Fig. S3), indicating that a portion of mitochondria were delivered to acidic lysosomes. Twenty-four hours of mitochondrial damage generated an overwhelming ratio of 561/458
nm signals in Parkin-expressing cells, indicating that most mitochondria were targeted by mitophagy (Fig. 5b, right region). Regarding the lung cancer-related Parkin mutations, the R275W and
N254S mutations produced partial effects compared with those of WT Parkin, while the A46T mutation blocked most of the mitophagic flux. TREATMENT WITH DFP PREVENTS THE SURVIVAL OF MUTANT
PARKIN-EXPRESSING LUNG CANCER CELLS According to previous reports, strong induction of mitophagy can trigger cancer cell death [39]. We found that long-term Parkin-mediated mitophagy
significantly induced cell death in lung cancer cells (Fig. 6a). However, expression of lung cancer-related Parkin mutants, especially the A46T mutant, resulted in decreased cell death
compared to that seen with WT Parkin (Fig. 6a). Interestingly and importantly, we found that DFP, an iron chelator and mitophagy inducer available for clinical use [21], significantly
increased the death of A46T Parkin-expressing cancer cells (Fig. 6b). Moreover, we found that in contrast to A/O treatment, DFP treatment did not affect mitochondrial dynamics and membrane
potential (Fig. S4), indicating that DFP was not toxic to mitochondrial function and that it can induce cancer cell death by increasing mitophagic signaling. Taken together, these data
suggest that induction of mitophagy may be a potential treatment for lung cancer caused by Parkin loss-of-function mutations. DISCUSSION Autophagy plays a fundamental role in regulating the
intracellular clearance of organelles, proteins and pathogens. Previous studies have shown that autophagy is tightly associated with a number of human diseases, including neurodegenerative
diseases, inflammatory diseases and cancer [14]. It is worth noting that studies of PINK1/Parkin-mediated mitophagy have mainly focused on neurodegenerative diseases, such as PD, whereas the
pathogenic relationship between PINK1/Parkin-mediated mitophagy and cancer development remains elusive. The current concept of mitophagy is that cells may use mitophagy to remove damaged
mitochondria to protect themselves against toxicity generated by damaged mitochondria, such as reactive oxygen species (ROS). However, emerging evidence indicates that long-term induction of
mitophagy in Parkin-expressing cells would lead to cell death through mechanisms currently unknown but possibly associated with apoptosis. For example, a previous report showed that
ceramide, a known tumor suppressor widely used in preclinical and clinical studies in cancer research, could trigger cancer cell death and tumor suppression through lethal mitophagy [39].
Consistent with this finding, we found that DFP treatment significantly decreased the survival of lung cancer cell lines expressing pathogenic Parkin mutants (Fig. 6b). One issue in the
field of autophagy is the lack of chemicals that can directly enhance downstream signaling in the pathway, such as inducers of autophagosome–lysosome fusion or autophagosome–substrate
recognition. Given that DFP treatment increased the number of autolysosome-targeted mitochondria and benefits of the research progress made in the pharmacological regulation of mitophagy
[40], our study not only is helpful for understanding the contribution of PINK1/Parkin-mediated mitophagy to the pathogenesis of lung cancer development but also offers experimental evidence
for the effective treatment of lung cancer and possibly other types of human cancers. Mechanistically, the A46T mutation may affect the conformation of the UBL domain of Parkin, thereby
either inhibiting the phosphorylation of UBL or blocking UBL release, which, in turn, stops the subsequent activation of the Parkin E3 catalytic center. In addition, the N254S and R275W
mutations are in the RING1 domain, which may inhibit the interaction between phosphorylated ubiquitin and the RING1 domain, thereby decreasing the E3 activity of Parkin. To date, it is not
understood why both PD and cancer, two entirely different diseases, can be caused by inactivating mutations in Parkin. Additionally, it is intriguing that the single mutation R275W in
_PARK2_ can either cause PD or cause cancer. There are several possibilities. First, non-cell-autonomous toxicity may also contribute to dopaminergic neuron degeneration in PD, since
germline mutation in _PARK2_ will result in loss of Parkin function across all cell types, including glial cells such as microglia, astrocytes and oligodendrocytes, in the central nervous
system. Second, various types of cellular stress in affected neurons or tumor cells may lead to different expression levels of mitophagy-related proteins. The combination of this variety and
Parkin dysfunction may cause different biological consequences; therefore, the R275W germline mutation, which has already been shown to impair mitophagy [41], is associated with both PD and
cancer. The last but perhaps the most important consideration is that dopaminergic neurons are more vulnerable when mitochondrial functions decline during aging. In support of this idea,
Parkin-null mice did not develop a neurodegenerative phenotype under normal conditions, whereas they exhibited dopaminergic neuron-specific degeneration when exposed to mitochondrial stress
[42]. Given that PD is a late-onset, age-related disease with increasing mitochondrial damage during disease progression, it is reasonable to postulate that Parkin-inactivating mutations
would accelerate the accumulation of damaged mitochondria accompanied by high levels of toxic species, such as ROS, eventually resulting in the death of dopaminergic neurons and the
development of PD during the aging process. Taken together, our findings suggest that both PD-linked and cancer-linked mutations in _PARK2_ can inhibit mitophagy in different ways, and
strategies to enhance functional mitophagy may be novel treatments for both diseases. REFERENCES * McWilliams TG, Muqit MM. PINK1 and Parkin: emerging themes in mitochondrial homeostasis.
Curr Opin Cell Biol. 2017;45:83–91. CAS PubMed Google Scholar * Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol. 2018;19:93–108.
CAS PubMed Google Scholar * Walden H, Muqit MM. Ubiquitin and Parkinson’s disease through the looking glass of genetics. Biochem J. 2017;474:1439–51. CAS PubMed PubMed Central Google
Scholar * Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci USA.
2011;108:16259–64. CAS PubMed Google Scholar * Fujiwara M, Marusawa H, Wang HQ, Iwai A, Ikeuchi K, Imai Y, et al. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene.
2008;27:6002–11. CAS PubMed Google Scholar * Picchio MC, Martin ES, Cesari R, Calin GA, Yendamuri S, Kuroki T, et al. Alterations of the tumor suppressor gene Parkin in non-small cell
lung cancer. Clin Cancer Res. 2004;10:2720–4. CAS PubMed Google Scholar * Bernardini JP, Lazarou M, Dewson G. Parkin and mitophagy in cancer. Oncogene. 2017;36:1315–27. CAS PubMed
Google Scholar * Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human
malignancies. Nat Genet. 2010;42:77–82. CAS PubMed Google Scholar * Xiong D, Wang Y, Kupert E, Simpson C, Pinney SM, Gaba CR, et al. A recurrent mutation in PARK2 is associated with
familial lung cancer. Am J Hum Genet. 2015;96:301–8. CAS PubMed PubMed Central Google Scholar * Yeo CW, Ng FS, Chai C, Tan JM, Koh GR, Chong YK, et al. Parkin pathway activation
mitigates glioma cell proliferation and predicts patient survival. Cancer Res. 2012;72:2543–53. CAS PubMed Google Scholar * Matsuda S, Nakanishi A, Minami A, Wada Y, Kitagishi Y.
Functions and characteristics of PINK1 and Parkin in cancer. Front Biosci (Landmark Ed). 2015;20:491–501. CAS Google Scholar * Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell.
2016;166:555–66. CAS PubMed PubMed Central Google Scholar * Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–64. CAS
PubMed Google Scholar * Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42. CAS PubMed Google Scholar * Wauer T, Simicek M,
Schubert A, Komander D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature. 2015;524:370–4. CAS PubMed PubMed Central Google Scholar * Kane LA, Lazarou M, Fogel AI, Li Y,
Yamano K, Sarraf SA, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–53. CAS PubMed PubMed Central Google Scholar *
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–14. CAS PubMed PubMed
Central Google Scholar * Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc
Natl Acad Sci USA. 2014;111:E4439–48. CAS PubMed Google Scholar * Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required
for efficient mitophagy. Proc Natl Acad Sci USA. 2016;113:E3349–58. CAS PubMed Google Scholar * Nezich CL, Wang C, Fogel AI, Youle RJ. MiT/TFE transcription factors are activated during
mitophagy downstream of Parkin and Atg5. J Cell Biol. 2015;210:435–50. CAS PubMed PubMed Central Google Scholar * Allen GF, Toth R, James J, Ganley IG. Loss of iron triggers
PINK1/Parkin-independent mitophagy. EMBO Rep. 2013;14:1127–35. CAS PubMed PubMed Central Google Scholar * Ying Z, Wang H, Fan H, Zhu X, Zhou J, Fei E, et al. Gp78, an ER associated E3,
promotes SOD1 and ataxin-3 degradation. Hum Mol Genet. 2009;18:4268–81. CAS PubMed Google Scholar * Ying Z, Wang H, Fan H, Wang G. The endoplasmic reticulum (ER)-associated degradation
system regulates aggregation and degradation of mutant neuroserpin. J Biol Chem. 2011;286:20835–44. CAS PubMed PubMed Central Google Scholar * Wang H, Ying Z, Wang G. Ataxin-3 regulates
aggresome formation of copper–zinc superoxide dismutase (SOD1) by editing K63-linked polyubiquitin chains. J Biol Chem. 2012;287:28576–85. CAS PubMed PubMed Central Google Scholar * Tao
Z, Wang H, Xia Q, Li K, Jiang X, Xu G, et al. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum Mol Genet.
2015;24:2426–41. CAS PubMed Google Scholar * Xia Q, Wang G, Wang H, Hu Q, Ying Z. Folliculin, a tumor suppressor associated with Birt–Hogg–Dube (BHD) syndrome, is a novel modifier of
TDP-43 cytoplasmic translocation and aggregation. Hum Mol Genet. 2016;25:83–96. CAS PubMed Google Scholar * Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, et al. TDP-43 loss of function
increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016;35:121–42. CAS PubMed Google Scholar * Zhou L, Wang HF, Ren HG, Chen D, Gao F, Hu QS, et al. Bcl-2-dependent
upregulation of autophagy by sequestosome 1/p62 in vitro. Acta Pharmacol Sin. 2013;34:651–6. CAS PubMed PubMed Central Google Scholar * Lv G, Sun D, Zhang J, Xie X, Wu X, Fang W, et al.
Lx2-32c, a novel semi-synthetic taxane, exerts antitumor activity against prostate cancer cells in vitro and in vivo. Acta Pharm Sin B. 2017;7:52–8. Google Scholar * Liu D, Tang H, Li XY,
Deng MF, Wei N, Wang X, et al. Targeting the HDAC2/HNF-4A/miR-101b/AMPK pathway rescues tauopathy and dendritic abnormalities in Alzheimer’s disease. Mol Ther. 2017;25:752–64. CAS PubMed
PubMed Central Google Scholar * Yang Y, Guan D, Lei L, Lu J, Liu JQ, Yang G, et al. H6, a novel hederagenin derivative, reverses multidrug resistance in vitro and in vivo. Toxicol Appl
Pharm. 2018;341:98–105. CAS Google Scholar * Wang X, Liu D, Huang HZ, Wang ZH, Hou TY, Yang X, et al. A novel microRNA-124/PTPN1 signal pathway mediates synaptic and memory deficits in
Alzheimer’s disease. Biol Psychiatry. 2018;83:395–405. CAS PubMed Google Scholar * Su Y, Deng MF, Xiong W, Xie AJ, Guo J, Liang ZH, et al. MicroRNA-26a/death-associated protein kinase 1
signaling induces synucleinopathy and dopaminergic neuron degeneration in Parkinson’s disease. Biol Psychiatry. 2019;85:769–81. CAS PubMed Google Scholar * Wu JC, Qi L, Wang Y, Kegel KB,
Yoder J, Difiglia M, et al. The regulation of N-terminal Huntingtin (Htt552) accumulation by Beclin1. Acta Pharmacol Sin. 2012;33:743–51. CAS PubMed PubMed Central Google Scholar * Ren
ZX, Zhao YF, Cao T, Zhen XC. Dihydromyricetin protects neurons in an MPTP-induced model of Parkinson’s disease by suppressing glycogen synthase kinase-3 beta activity. Acta Pharmacol Sin.
2016;37:1315–24. CAS PubMed PubMed Central Google Scholar * Fang LM, Li B, Guan JJ, Xu HD, Shen GH, Gao QG, et al. Transcription factor EB is involved in autophagy-mediated
chemoresistance to doxorubicin in human cancer cells. Acta Pharmacol Sin. 2017;38:1305–16. CAS PubMed PubMed Central Google Scholar * Sun N, Malide D, Liu J, Rovira II, Combs CA, Finkel
T. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat Protoc. 2017;12:1576–87. CAS PubMed Google Scholar * Chen Y, Xu S, Wang N, Ma Q, Peng
P, Yu Y, et al. Dynasore suppresses mTORC1 activity and induces autophagy to regulate the clearance of protein aggregates in neurodegenerative diseases. Neurotox Res. 2019; in press.
https://doi.org/10.1007/s12640-019-00027-9. CAS PubMed Google Scholar * Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, et al. Ceramide targets autophagosomes to
mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8:831–8. CAS PubMed PubMed Central Google Scholar * Georgakopoulos ND, Wells G, Campanella M. The pharmacological
regulation of cellular mitophagy. Nat Chem Biol. 2017;13:136–46. CAS PubMed Google Scholar * Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for
Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–106. CAS PubMed PubMed Central Google Scholar * Pickrell AM, Huang CH,
Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, et al. Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron.
2015;87:371–81. CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the National Natural Science Foundation of China (Nos. 31771117,
31701222 and 31571053), the National Key Plan for Scientific Research and Development of China (No. 2017YFC0909100), a Project Funded by Jiangsu Key Laboratory of Neuropsychiatric Diseases
(BM2013003) and a Project Funded by the Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD). AUTHOR INFORMATION Author notes * These authors contributed
equally: Zeng-li Zhang, Na-na Wang AUTHORS AND AFFILIATIONS * Department of Respiration, The Second Affiliated Hospital of Soochow University, Suzhou, 215004, China Zeng-li Zhang &
Min-hua Shi * Jiangsu Key Laboratory of Neuropsychiatric Diseases and College of Pharmaceutical Sciences, Soochow University, Suzhou, 215123, China Na-na Wang, Qi-lian Ma, Yang Chen, Li Yao,
Hong-feng Wang & Zheng Ying * School of Pharmacy, Key Laboratory of Molecular Pharmacology and Drug Evaluation (Yantai University), Ministry of Education, Yantai University, Yantai,
264005, China Zheng Ying * Jiangsu Key Laboratory of Preventive and Translational Medicine for Geriatric Diseases, College of Pharmaceutical Sciences, Soochow University, Suzhou, 215021,
China Zheng Ying * Key Laboratory of Nuclear Medicine, Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi, 214063, China Li
Zhang * National University of Singapore (Suzhou) Research Institute, Suzhou, 215123, China Qiu-shi Li Authors * Zeng-li Zhang View author publications You can also search for this author
inPubMed Google Scholar * Na-na Wang View author publications You can also search for this author inPubMed Google Scholar * Qi-lian Ma View author publications You can also search for this
author inPubMed Google Scholar * Yang Chen View author publications You can also search for this author inPubMed Google Scholar * Li Yao View author publications You can also search for this
author inPubMed Google Scholar * Li Zhang View author publications You can also search for this author inPubMed Google Scholar * Qiu-shi Li View author publications You can also search for
this author inPubMed Google Scholar * Min-hua Shi View author publications You can also search for this author inPubMed Google Scholar * Hong-feng Wang View author publications You can also
search for this author inPubMed Google Scholar * Zheng Ying View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS ZY, HFW, MHS and QSL designed
the experiments and drafted the manuscript; ZLZ, NNW, QLM, YC, LY and LZ performed the experiments. CORRESPONDING AUTHORS Correspondence to Hong-feng Wang or Zheng Ying. ETHICS DECLARATIONS
COMPETING INTERESTS The authors declare no competing interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE
THIS ARTICLE Zhang, Zl., Wang, Nn., Ma, Ql. _et al._ Somatic and germline mutations in the tumor suppressor gene _PARK2_ impair PINK1/Parkin-mediated mitophagy in lung cancer cells. _Acta
Pharmacol Sin_ 41, 93–100 (2020). https://doi.org/10.1038/s41401-019-0260-6 Download citation * Received: 22 March 2019 * Accepted: 21 May 2019 * Published: 08 July 2019 * Issue Date:
January 2020 * DOI: https://doi.org/10.1038/s41401-019-0260-6 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * autophagy * mitophagy * Parkin *
ubiquitin * cancer
Trending News
Small doses of alcohol affect driving skills | science newsScience News was founded in 1921 as an independent, nonprofit source of accurate information on the latest news of scien...
Waste-to-biofuels technology ready for global rolloutBreakthrough technology developed by Enerkem converts a multitude of waste material into renewable fuel and chemicals, m...
Drought dries up copper canyon waterfall although some blame miningA waterfall in the Copper Canyon in Ocampo, Chihuahua, has dried up due to the severe drought affecting the area. The 24...
5 signs a house is a money pitThe spring home-buying season is in full swing, and if forecasts are correct it’s going to be a busy one. Despite mortga...
The bcl-2 pro-survival protein a1 is dispensable for t cell homeostasis on viral infectionABSTRACT The physiological role of the pro-survival BCL-2 family member A1 has been debated for a long time. Strong mRNA...
Latests News
Somatic and germline mutations in the tumor suppressor gene park2 impair pink1/parkin-mediated mitophagy in lung cancer cellsABSTRACT _PARK2_, which encodes Parkin, is a disease-causing gene for both neurodegenerative disorders and cancer. Parki...
Texas school districts face safety dilemma after governor lifts mask orderTexas school leaders found themselves once again at the center of a COVID-19 safety debate after Gov. Greg Abbott lifted...
Pbks vs srh ipl 2024 match today: live streaming, telecast & playing 11 detailsPunjab Kings (PBKS) and Sunrisers Hyderabad (SRH) will clash today on Tuesday, 9 April 2024, in the match 23 of the Indi...
Bournemouth know lloyd kelly admires eddie howe with newcastle ready to make bidEddie Howe is now ready to test Bournemouth’s resolve and step up his interest in signing Lloyd Kelly, who the Cherries ...
Carnegie Endowment for International Peace | Carnegie Endowment for International PeaceGlobal LocationsresearchemissaryaboutexpertsmoresupportprogramseventsblogspodcastsvideosNewslettersAnnual Reportscareers...