Novel pathogenic gata6 variant associated with congenital heart disease, diabetes mellitus and necrotizing enterocolitis
Novel pathogenic gata6 variant associated with congenital heart disease, diabetes mellitus and necrotizing enterocolitis"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT BACKGROUND Pathogenic _GATA6_ variants have been associated with congenital heart disease (CHD) and a spectrum of extracardiac abnormalities, including pancreatic agenesis,
congenital diaphragmatic hernia, and developmental delay. However, the comprehensive genotype-phenotype correlation of pathogenic _GATA6_ variation in humans remains to be fully understood.
METHODS Exome sequencing was performed in a family where four members had CHD. In vitro functional analysis of the _GATA6_ variant was performed using immunofluorescence, western blot, and
dual-luciferase reporter assay. RESULTS A novel, heterozygous missense variant in _GATA6_ (c.1403 G > A; p.Cys468Tyr) segregated with affected members in a family with CHD, including
three with persistent truncus arteriosus. In addition, one member had childhood onset diabetes mellitus (DM), and another had necrotizing enterocolitis (NEC) with intestinal perforation. The
p.Cys468Tyr variant was located in the c-terminal zinc finger domain encoded by exon 4. The mutant protein demonstrated an abnormal nuclear localization pattern with protein aggregation and
decreased transcriptional activity. CONCLUSIONS We report a novel, familial _GATA6_ likely pathogenic variant associated with CHD, DM, and NEC with intestinal perforation. These findings
expand the phenotypic spectrum of pathologic _GATA6_ variation to include intestinal abnormalities. IMPACT * Exome sequencing identified a novel heterozygous _GATA6_ variant (p.Cys468Tyr)
that segregated in a family with CHD including persistent truncus arteriosus, atrial septal defects and bicuspid aortic valve. Additionally, affected members displayed extracardiac findings
including childhood-onset diabetes mellitus, and uniquely, necrotizing enterocolitis with intestinal perforation in the first four days of life. * In vitro functional assays demonstrated
that _GATA6_ p.Cys468Tyr variant leads to cellular localization defects and decreased transactivation activity. * This work supports the importance of _GATA6_ as a causative gene for CHD and
expands the phenotypic spectrum of pathogenic _GATA6_ variation, highlighting neonatal intestinal perforation as a novel extracardiac phenotype. You have full access to this article via
your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS THE C.1617DEL VARIANT OF _TMEM260_ IS IDENTIFIED AS THE MOST FREQUENT SINGLE GENE DETERMINANT FOR JAPANESE PATIENTS WITH
A SPECIFIC TYPE OF CONGENITAL HEART DISEASE Article Open access 26 February 2024 IDENTIFICATION AND CHARACTERIZATION OF A NOVEL _ELN_ MUTATION IN CONGENITAL HEART DISEASE WITH PULMONARY
ARTERY STENOSIS Article Open access 08 July 2021 EXPANDING THE PHENOTYPIC SPECTRUM OF _NOTCH1_ VARIANTS: CLINICAL MANIFESTATIONS IN FAMILIES WITH CONGENITAL HEART DISEASE Article Open access
22 May 2024 INTRODUCTION The GATA family of transcription factors encode for zinc finger DNA binding proteins that play critical regulatory roles in organogenesis.1 Among this family,
GATA4, GATA5, and GATA6 have been implicated in cardiovascular morphogenesis and expressed in overlapping but distinct spatiotemporal patterns during development.2,3 Pathogenic variation in
_GATA4_, _GATA5_ and _GATA6_ have been reported in a spectrum of human congenital heart disease (CHD).4,5 Pathogenic variants in _GATA4_ were first reported in individuals with cardiac
septal defects including atrial septal defects (ASD), ventricular septal defects (VSD), and atrioventricular septal defects (AVSD).6 The CHD phenotypes associated with pathologic _GATA4_
variants have expanded to include pulmonary stenosis (PS), tetralogy of Fallot (TOF) and bicuspid aortic valve (BAV).7,8,9,10,11 The primary cardiac phenotype associated with _GATA5_
pathogenic variants is BAV although additional phenotypes include VSD, ASD, TOF and double outlet right ventricle (DORV).3,12,13,14,15,16 The expression pattern of GATA6 during embryogenesis
partially overlaps with GATA4 and GATA5 in the heart and other tissues, including the digestive system and the extraembryonic endoderm.17,18,19 In particular, GATA6 plays a role in the
developing second heart field (SHF) and in the recruitment of cardiac neural crest cells, which together form the cardiac outflow tract.20 _GATA6_ variants in humans were first discovered in
individuals with persistent truncus arteriosus (PTA).21 Subsequently, _GATA6_ pathogenic variants have been identified in other malformations of cardiac outflow tract defect, including TOF,
pulmonary atresia with ventricular septal defect (PA/VSD), DORV and transposition of the great arteries (TGA).22,23,24,25,26,27 Similar to GATA4 and GATA5, _GATA6_ variants have been found
in individuals with a variety of CHD phenotypes including ASD, VSD, PS, BAV and patent ductus arteriosus (PDA).28,29,30 Unique from _GATA4_ and _GATA5_, _GATA6_ pathogenic variants are found
to be associated with extracardiac abnormalities in humans. Consistent with this, GATA6 plays a critical role for regulating gene expression in the development of endoderm from which the
pancreas and gut are derived.18,19 Pancreatic agenesis was initially identified as an extracardiac phenotype in individuals with CHD who had _GATA6_ variants.31 Pancreatic abnormalities are
the most common extracardiac features associated with _GATA6_ pathogenic variation, which range from severe hypoplasia or agenesis of the pancreas with neonatal or childhood-onset diabetes
mellitus (DM) to a mild phenotype such as adult-onset DM.32,33,34,35 Additional extracardiac malformations have also been reported, including hepatobiliary and gastrointestinal
abnormalities.31,32,36,37 Gastrointestinal abnormalities associated with _GATA6_ variants include congenital diaphragmatic hernia, but evidence regarding the association between _GATA6_
pathogenic variation and congenital anomalies affecting gut such as small intestine, colon and rectum is less common.37 Accordingly, the broad phenotypic spectrum and genotype-phenotype
correlations for genetic variation in _GATA6_ for cardiac and extracardiac diseases remains to be defined. Here, we identified a novel, heterozygous missense variant in _GATA6_ that
segregated with disease in a family with a spectrum of CHD and extracardiac phenotypes using exome sequencing. The father had PTA while three children also had CHD, including PTA, ASD, BAV
and interrupted aortic arch. In addition, the father had childhood-onset DM and one child had necrotizing enterocolitis (NEC) with intestinal perforation. Using in vitro assays, we
demonstrate that the GATA6 mutant protein displays functional deficits in nuclear localization and reduced transcriptional activity. This work highlights the phenotypic spectrum for
_GATA6_-associated disease from CHD and DM to even gastrointestinal abnormalities. METHODS ETHICS STATEMENT Family members were recruited under an approved Nationwide Children’s Hospital
Institutional Review Board protocol (#IRB09-00339). Written informed consent was obtained from study subjects 18 years of age and older and from parents for study subjects under 18 years of
age, with assent obtained from subjects 9-17 years of age. DNA ISOLATION Blood samples or saliva samples (Oragene) were obtained and DNA was isolated using Gentra Puregene Kit (Qiagen,
Hilden, Germany) as previously described.38 EXOME LIBRARY CONSTRUCTION AND EXOME SEQUENCING Exome libraries were constructed using Agilent Clinical Research Exome Kit v1 (Agilent
Technologies, CA). Paired-end 150 base pair reads were generated for exome-enriched libraries sequenced on Illumina NovaSeq to a targeted depth of 100x coverage. EXOME SEQUENCING DATA
PIPELINE We analyzed and annotated exome sequencing data using methods as previously described.38 VARIANT ANALYSIS Variant filtering on frequency, effect, and predicted impact, and
segregation with disease was performed as described in Manivannan et al. 2020.39 _GATA6_ genomic region flanking the identified variant was amplified using primers GATA6.gen.For and
GATA6.gen.Rev (Supplementary Table S1) to confirm the presence of the variant using Sanger sequencing method. The novel _GATA6_ variant identified was assessed for pathogenicity using the
American College of Medical Genetics and Genomics-Association for Molecular Pathology (ACMG/AMP) guidelines.40 The predicted three-dimensional structure of the mutant protein was created
using Missense3D.41 The wildtype and mutant protein structures were visualized and attribution of the residue position to the protein function was analyzed using Jmol
(https://jmol.sourceforge.net/). Gibbs free energy calculations were performed using DynaMut2.42 PLASMID CONSTRUCTION Full length human _GATA6_ cDNA (Refseq ID NM_005257) was obtained from
Harvard Plasmid Database. _GATA6_ coding region with the 3’UTR sequence was amplified using PCR with primers FP.GATA6.BamHI and RP.GATA6.EcoRI (Supplementary Table S1), and cloned into
_BamHI/EcoRI_ site in the pIRES-mCherry plasmid (a gift from Ellen Rothenberg, Addgene plasmid # 80139). Then, _GATA6_-coding region, 3ʹUTR and IRES mCherry were excised using _BamHI/EcoRI_
and introduced in frame with the EGFP tag in a modified pEGFP-C1 plasmid (Clontech) to generate the pEGFP-GATA6-IRES-mCherry plasmid. When transfected, this construct leads to the production
of an EGFP tagged GATA6 protein and independently translated mCherry from the same transcript. The EGFP tag distinguishes the overexpressed cDNA from _GATA6_, while the independently
produced mCherry acts as a readout of mRNA transcription and also serves as a transfection control allowing direct comparison of cellular levels of overexpressed GATA6 protein variant. To
generate the variant, site-directed mutagenesis was used with pEGFP-GATA6-IRES-mCherry as the template and using the primers FP.GATA6.C468Y and RP.GATA6.C468R (Supplementary Table S1), and
the Agilent Quickchange II kit.43 All vectors constructed were verified by sequencing. CELL CULTURE HEK293 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 4.5 g/L
Glucose, 4 mM L-Glutamine, 1 mM sodium pyruvate, and 1.5 g/L sodium bicarbonate (ATCC 30–2002), supplemented with 10% fetal bovine serum, 100 I.U./mL penicillin and 100 (μg/mL) streptomycin
at 37 °C incubator with 5% CO2. Cells were transfected with 2 μg of the plasmid with Lipofectamine 3000 reagent (Thermo Fisher Scientific, #L3000015) with Opti-MEM medium (Thermo Fisher
Scientific, #31985-070) according to manufacturer’s recommendations. Transfection media was removed five hours post-transfection and replaced by normal growth media. Cells were collected for
western blotting or immunofluorescence analysis 48 h after transfection. WESTERN BLOTTING Cell lysates were prepared from cultured HEK293 cells using RIPA Lysis and Extraction Buffer
(ThermoFisher Scientific, #89900) supplemented with Halt Protease Inhibitor Cocktail (ThermoFisher Scientific, #87785). BCA Protein Assay Kit (ThermoFisher Scientific, #23227) was used to
estimate protein concentration. Cell lysates were mixed with 6X Laemmli SDS-Sample Buffer (Boston BioProducts, #BP-111R) containing β-mercaptoethanol and boiled for 5 min. Protein samples
were separated in 4 to 20% Mini-PROTEAN TGX Precast Gels (Bio-Rad, #4561094), transferred into a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, #1620177) and blocked with 5% nonfat milk
in Tris-Buffered Saline containing 0.1% Tween 20 (TBST). Membranes were probed with primary antibodies against GATA6 (1:500, R&D Systems, #AF1700), GAPDH (1:1000, Novus Biologicals,
#NB300-221). After probing with primary antibodies, membranes were further probed with horseradish peroxidase–conjugated anti-rabbit and anti-mouse secondary antibodies (Vector Laboratories,
#PI-1000 and PI-2000). Western blots were developed using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, #32106). Restore Western Blot Stripping Buffer (Thermo Fisher
Scientific, #21059) was used for re-probing with different primary antibodies following the manufacturer’s protocol. Protein levels were quantified by densitometric analysis using ImageJ
software and normalized to GAPDH. LUCIFERASE TRANSACTIVATION ASSAYS HEK293 cells were transiently transfected using Lipofectamine 3000 reagent (Thermo Fisher Scientific, #L3000015) with 150
ng of atrial natriuretic factor (ANF) luciferase reporter and with 300 ng of GATA6 wildtype or GATA6 p.Cys468Tyr, using previously described methods.15,23 Luciferase activity was measured 48
h after transient transfection according to the manufacturer’s instructions (Promega, Madison, WI). The luciferase signal was normalized to the _Renilla_ signal to control for variation in
transfection efficiency. Three independent experiments were performed in triplicate. IMMUNOFLUORESCENCE Cultured HEK293 cells were fixed with 2.5% PFA 48 h after transient transfection and
permeabilized with Phosphate-Buffered Saline Triton (PBST, PBS containing 0.1% TritonX100). After permeabilization, non-specific immunoreactions were blocked using 1% BSA in PBST for 1 h at
room temperature and incubated overnight with primary antibodies: Chicken anti-GFP (1:1000, Abcam, #ab13970) and Rabbit anti-mCherry (1:1000, Abcam, #ab167453). After primary antibody
incubation, cells were washed with PBST and incubated with anti-chicken and anti-rabbit secondary antibodies conjugated to Alexa Fluor 488 and 568 (ThermoFisher Scientific, #A11039 and
#A11011) for 1 h in the dark. Nuclei were stained with DAPI (1.5 μg/ml; Sigma-Aldrich, #D9542). Keyence BZ-X800 Fluorescence microscope was used to capture images. Expression of EGFP
tagged-GATA6 wildtype or GATA6 p.Cys468Tyr protein and protein aggregation was quantified using ImageJ by counting the number of particles by counting the nuclei (DAPI) in the same image, as
previously described.44 STATISTICAL ANALYSIS All experiments were performed at least in triplicate. Statistical analysis was performed in the GraphPad Prism 9 software package using
Two-tailed Student’s _t_ test. _P_ values of ≤0.05 were considered as statistically significant. RESULTS FAMILY WITH MULTIPLE MEMBERS AFFECTED WITH CONGENITAL HEART DISEASE We identified a
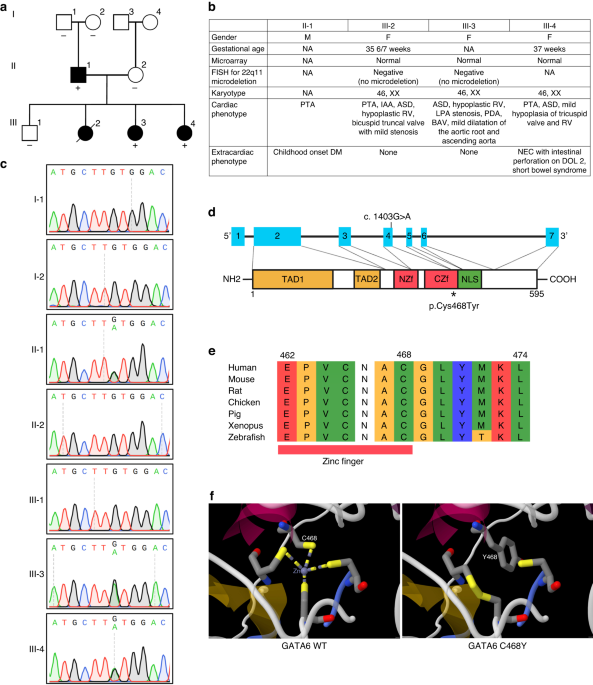
family where four members displayed CHD along with additional non-cardiac diseases (Fig. 1a). The father had PTA and childhood-onset DM. Among the four children, three had CHD. Two children
had PTA while another ASD, BAV and mild right ventricle (RV) hypoplasia (Fig. 1b). The proband (III-4) was prenatally diagnosed with PTA (Collett and Edwards type II)45 due to the family
history of CHD in the father. She was born at 37 weeks of gestation to a 28 year old mother. The birthweight was 2580 g with Apgar scores of 7 and 8. On the first day of life, she was
transferred to the cardiothoracic intensive care unit (CTICU) for the management of PTA. In addition to PTA, ASD, mild hypoplasia of tricuspid valve and RV were noted by echocardiogram and
later demonstrated by cardiac MRI. She was made nil per os (NPO) on admission but allowed limited self-advance feedings with breastmilk. She was hemodynamically stable breathing in room air,
with adequate near-infrared spectroscopy and diastolic blood pressures without cardiovascular or respiratory compromise. However, at 44 h after birth, a routine morning X-ray showed
pneumatosis, dilated bowel loops and portal venous gas. Clinically, she also developed abdominal distension and was diagnosed with NEC. She was made NPO and piperacillin/tazobactam was
started. At four days of age, an exploratory laparotomy revealed necrotic bowel of the distal ileum and distal right colon along with perforation of the proximal transverse colon. She
underwent bowel resection, and end ileostomy was performed in the right lower quadrant with the mesentery of the small bowel facing medially. Surgical pathology report revealed marked
mucosal necrosis and acute inflammation with regions of full-thickness bowel necrosis in the ileum, right colon and proximal transverse colon. After recovering from NEC, she underwent
bilateral pulmonary artery (PA) banding on 40 days of age. She then underwent ileostomy takedown and anastomosis for restoration of continuity, and gastrostomy tube placement at 65 days of
age. Subsequently, she developed complete colonic obstruction requiring multiple surgeries, including an emergent exploratory laparotomy (99 days old) and recreation of ileostomy (105 days
old). She was discharged from the hospital at 181 days of age. She underwent complete repair of PTA with VSD closure, utilizing a 12 mm Hancock valved conduit as the right ventricular to
pulmonary artery conduit, patch augmentation of both pulmonary arteries and ASD closure at the age of nine months without complication and was discharged in six days. Due to NEC and its
complications, she developed short bowel syndrome and was able to achieve enteral autonomy by the age of 15 months. Subsequently, she underwent ostomy takedown and ileocolonic reanastomosis
creation to increase bowel length at 19 months of age. She recovered from the operation and the clinical course was uneventful for one year. Of note, the tricuspid valve remains mildly
hypoplastic while the mild RV hypoplasia resolved by echocardiogram at four years of age. She had normal karyotype of 46, XX, normal results of 22q11 FISH and SNP microarray. The father
(II-1), a 35 year old man, was diagnosed with PTA (Collett and Edwards type II) and at 3 months of age underwent complete repair of PTA with closure of ASD. Furthermore, he was diagnosed
with DM at the age of 14 (by report) and is insulin-dependent. The sibling (III-2) was diagnosed with PTA with interrupted aortic arch (type B) and bicuspid truncal valve with mild stenosis
at birth due to cyanosis. In addition, there was hypoplasia of the tricuspid valve, RV, and proximal branch pulmonary arteries along with a large ASD. She died at two weeks of age from
circulatory failure at another institution. She had normal karyotype of 46, XX, normal results of 22q11 FISH and SNP microarray. The other sibling (III-3) was diagnosed with large ASD, PDA
and mild hypoplasia of tricuspid valve, RV, and pulmonary valve at birth due to heart murmur and family history of CHD. In addition, left pulmonary artery (LPA) hypoplasia with associated
proximal LPA stenosis was noted. At six months of age, she underwent implantation of LPA stent and PDA occlusion with Amplatzer Vascular Plug II. She was then diagnosed with supraventricular
tachycardia. Subsequently, electrophysiology study showed atrioventricular nodal reentrant tachycardia (AVNRT) and she underwent repeated LPA stent dilation. Given her arrhythmia, she did
not undergo ASD closure. Her recent echocardiogram demonstrated moderate RV dilation and bidirectional shunting through a moderate-sized ASD with her baseline oxygen saturations of 93% at
the age of eight years. Notably, BAV with mild aortic valve insufficiency and no aortic valve stenosis as well as mild to moderate dilatation of the aortic root and ascending aorta were also
detected. She is being followed to determine the plan for ASD closure. Her chromosome analysis showed a normal female karyotype of 46, XX and FISH result showed no deletion of 22q11.
IDENTIFICATION OF NOVEL MISSENSE VARIANT IN GATA6 BY EXOME SEQUENCING Due to the multiple family members affected with CHD, exome sequencing was performed on the proband-parent trio (III-4,
II-1, II-2) as well as the affected sibling (III-3), the unaffected sibling (III-1) and unaffected paternal grandparents (I-1 and I-2). Sequencing data were analyzed using our previously
published pipeline, Churchill, for calling variants.46 Variants were prioritized using minor allele frequency (MAF) (≤0.001) across population in gnomAD, sequencing quality filtering,
predicted functional impact, and segregation of candidate variants (Supplementary Fig. S1).47,48,49,50,51,52,53,54 This approach resulted in the identification of a single heterozygous
missense variant in _GATA6_ (NM_005257.5: c.1403 G > A; p.Cys468Tyr) that segregated in the family, i.e. present in affected family members and absent from unaffected members. Therefore,
we focused on the _GATA6_ variant since it was the only candidate disease-causing variant in the family. We confirmed presence of the heterozygous _GATA6_ variant in the proband (III-4),
affected father (II-1), and affected sibling (III-3) and its absence in the unaffected mother (II-2) and unaffected sibling (III-1) by Sanger sequencing (Fig. 1c). Further, testing of
paternal grandparents (I-1 and I-2) demonstrated that this was a de novo variant in the father II-1 (Fig.1c). This _GATA6_ variant was not found in control populations in gnomAD database.
The variant in _GATA6_ was rated likely pathogenic according to ACMG/AMP guidelines (Table 1). The _GATA6_ variant occurs within the C-terminal zinc finger (ZF) domain (Fig. 1d).
Cross-species alignment of GATA6 protein demonstrates highly conserved cysteine at position 468 (Fig. 1e). Three-dimensional protein structure of C-terminal ZF in GATA6 showed that the
structure introduced by ZF being bound to these four cysteines is imperative to proper functioning in GATA6 wildtype protein. Substitution of the cysteine at codon 468 with a tyrosine
changes the ZF structure from four closed coordination sphere to three open coordinate system (Fig. 1f). This structural modeling suggests that _GATA6_ p.Cys468Tyr variant likely severely
impacts the stability of the zinc finger. IN VITRO _ANALYSIS OF THE GATA6_ P.CYS468TYR VARIANT To assess the structural and functional impact of _GATA6_ variant, we generated an EGFP-tagged
human _GATA6_ cDNA construct that also expresses mCherry, allowing for simultaneous evaluation of mRNA stability and protein stability (Fig. 2a). Site-directed mutagenesis was performed on
this construct to introduce the _GATA6_ p.Cys468Tyr variant. Using this construct, we tested the stability of the _GATA6_ p.Cys468Tyr variant. Expression constructs of _GATA6_ wildtype and
_GATA6_ p.Cys468Tyr variant were transiently transfected into HEK293 cells. Immunofluorescence analysis of EGFP-tagged _GATA6_ constructs that overexpress mCherry from the same transcript
after transfection in HEK293 cells demonstrated that the GATA6 wildtype localized to the nucleus, whereas the GATA6 p.Cys468Tyr variant exhibited an abnormal localization pattern in the
nucleus (Fig. 2b). There was no significant difference in the production of mCherry between the wildtype and variant constructs, suggesting that the transfection and transcription of the
GATA6 variant construct was comparable to the wildtype GATA6 construct (Fig. 2b). To identify and quantify protein aggregates, the number of particles divided by the number of nuclei was
counted. The number of particles/nucleus was significantly increased in GATA6 p.Cys468Tyr variant as compared to GATA6 wildtype, suggesting aggregation of GATA6 p.Cys468Tyr protein (Fig.
2c). Western blot analyses showed reduced protein expression in GATA6 p.Cys468Tyr variant protein compared to GATA6 wildtype protein (Fig. 2d). In transient transfection transactivation
assays using the GATA6-dependent cardiac enhancer, ANF, upstream of a luciferase reporter, the GATA6 p.Cys468Tyr mutant protein demonstrated significantly decreased transcriptional activity
when compared with wildtype GATA6 (Fig. 2e). Together, these in vitro functional analyses indicated that _GATA6_ p.Cys468Tyr variant leads to functional deficits of the GATA6 protein and
likely impacts the regulation of downstream target genes during heart development. DISCUSSION Here, we investigated the genetic etiology of a familial case of CHD, predominantly PTA. Using
an exome sequencing approach, we identified a novel, heterozygous missense _GATA6_ variant (p.Cys468Tyr) which segregated with affected family members. In vitro functional analyses
demonstrated that _GATA6_ p.Cys468Tyr variant leads to cellular localization defects and decreased transactivation activity when compared with wildtype _GATA6_. While the spectrum of disease
phenotypes found in family members harboring the likely pathogenic variant included CHD and childhood onset DM, as reported by others,28 it also includes NEC and intestinal perforation
within the first week of life. These findings support the importance of _GATA6_ as a causative gene for CHD and highlights a novel extracardiac phenotype, early NEC, that can be associated
with _GATA6_ variants. This study will contribute to the growing literature about the association of pathogenic GATA6 variation with intestinal abnormalities as well as cardiac and
pancreatic malformations (Fig. 3). _GATA6_ variants have been identified in human CHD.55 The prevalence of _GATA6_ variants ranged from 0.5-2.0% in non-familial CHD in initial reports with a
higher frequency in cardiac outflow tract defects such as PTA and TOF.21,22,23,24,25,26,27,56 Recent exome sequencing studies in a large CHD cohort with more than 4000 CHD patients
conducted by the Pediatric Cardiac Genome Consortium (PCGC) discovered 9 heterozygous de novo variants in _GATA6_ including loss of function (LoF) and damaging missense variants.57,58 CHD
associated with _GATA6_ pathogenic variants is mainly cardiac outflow tract malformations, such as PTA, TOF and DORV, along with septal defects in some patients (Supplementary Table S2). In
addition, approximately 40% of these CHD patients with _GATA6_ variants also had extracardiac phenotypes, including pancreatic agenesis, congenital diaphragmatic hernia, and
neurodevelopmental disorders. Approximately 25% of previously reported _GATA6_ missense variants associated with CHD were located in the C-terminal DNA-binding zinc finger domain encoded by
exon 4 (amino acids 435-477). Interestingly, pancreatic malformations and congenital diaphragmatic hernia were observed in 9 out of 11 CHD patients who had _GATA6_ missense mutations within
exon 4 that encoding the C-terminal DNA-binding zinc finger domain (amino acids 435-477), whereas these extracardiac abnormalities were not found in any of the 32 patients with _GATA6_
missense variants in the other exons.59 Consistent with these findings, the novel _GATA6_ missense variant (p.Cys468Tyr) identified in our study was located in the C-terminal zinc finger
domain encoded by exon 4, leading to both cardiac and extracardiac phenotypes. The cysteine at codon 468 in the C-terminal zinc finger domain is highly conserved among the GATA family
members and plays an important role in the formation of the core zinc module for the GATA binding site recognition and protein interactions.60,61 The p.Cys468Tyr variant presumably disrupts
the core zinc module, leading to defects in the DNA binding as well as interactions with transcription factors, which thus causes dysregulation of GATA6 downstream target genes implicated in
multiple cell lineages mainly associated with cardiovascular and digestive organs. This works highlights early NEC as a new gastrointestinal phenotype associated with _GATA6_ variants. To
the best of our knowledge, two cases with CHD and intestinal abnormalities associated with _GATA6_ exon 4 missense mutations (p.Thr452Ala and p.Asn466Asp) and one case with _GATA6_ LoF
mutation were previously reported (Fig. 3), although NEC was not observed in either case.31,62 Remarkably, the proband developed NEC with intestinal perforation on the second day of life
without evidence of circulatory failure. NEC has been described in PTA with pulmonary overcirculation, which typically becomes clinically apparent over the first few days to weeks of life as
the pulmonary vascular resistance decreases.63 The pathophysiology of NEC is multifactorial, with premature birth being a primary determining factor as it is almost exclusively found in
preterm infants and its incidence is inversely correlated to gestational age at birth.64,65 Other risk factors associated with the development of NEC include bacterial infection, maternal
cocaine use, hypoxia and CHD primarily single ventricle physiology and left ventricular outflow tract obstructive lesions and episodes of systemic perfusion failure or shock.66,67,68 The
proband in our study had PTA, but did not have any of these risk factors including prematurity or episodes of systemic circulatory failure. Our institutional feeding protocol considers
respiratory status, respiratory support, near-infrared spectroscopy and diastolic blood pressures, which are surrogate markers for end organ or gut perfusion. Accordingly, we postulate that
NEC with intestinal perforation in the first four days of life in this patient is part of the _GATA6_ extracardiac phenotypic spectrum, although we cannot rule out the possibility that the
more severe CHD phenotype might influence the onset of NEC when compared to other reported cases with _GATA6_ exon 4 missense variants that did not have NEC (Fig. 3). The molecular
mechanisms how the _GATA6_ mutations cause PTA have been well investigated using genetically engineered mouse models.20 GATA6 is critical for the development of SHF and in the recruitment of
cardiac neural crest cells, contributing to the formation of the cardiac outflow tract by regulating semaphorin 3 C (_SEMA3C_) and plexin A2 (_PLXNA2_) during development.19,20,21 Further,
a molecular network involving _GATA6_, _FOXC1/2_, _TBX1_, _SEMA3C_, and _FGF8_ play important roles in the interaction between SHF and cardiac neural crest cells, and the failure of this
interaction results in PTA.69,70 Our study showed an abnormal localization pattern and decreased transcriptional activity of a novel GATA6 p.Cys468Tyr mutant protein that results in PTA
likely due to the disruption of the above molecular network during cardiovascular development. We also demonstrated protein aggregates of a novel GATA6 p.Cys468Tyr mutant protein. Protein
aggregates have been studied as causes underlying contractile dysfunction and human cardiomyopathy associated with genetic mutations, including _RBM20_, desmins, αB-crystallin, and COP9
signalosome subunit 8 (CSN8).71,72,73,74 In addition, a TBX1 mutant protein identified in a patient with TOF showed nuclear and cytoplasmic aggregates in transfected cells.75 However, the
mechanisms by which protein aggregates of genetic variants contribute to CHD in cardiac development remain unclear. Further studies are warranted to investigate the role of protein
aggregates underlying CHD phenotypes associated with _GATA6_ variants. The molecular mechanisms by which _GATA6_ variants cause NEC has not been explored. GATA6 is known to play a role in
intestinal development as conditional deletion in the developing intestine results in loss of the normal architecture of the intestinal villus with altered cell populations.76,77,78 While
intestinal abnormalities associated with _GATA6_ pathologic variants have been reported, including intestinal malrotation as well as protein losing enteropathy,28,79 the genetics of NEC
remains largely unknown.80 Large sequencing-based pilot studies have identified _NOD2_ (nucleotide-binding oligomerization domain containing 2), inducing autophagy pathway, and _SIGIRR_
(single immunoglobulin interleukin-1 related receptor), a modulator of Toll-like receptor-interleukin 1 receptor signaling, as potential loci for NEC.81,82 Since excessive inflammation is a
feature of NEC, several pro-inflammatory and cytokine genes have also been investigated, showing variable results.83 Additionally, the expression of tight junction proteins-coding genes such
as zonula occludens-1 (ZO-1) is shown to be significantly downregulated in intestinal specimens from NEC patients and animal models.84,85 Recently, investigators demonstrated that
conditional deletion of _Gata6_ in the gut epithelium significantly impacted intestinal barrier integrity and decreased expression of ZO-1, leading to enhanced intestinal permeability and
susceptibility to gut inflammation. Further, GATA6 directly modulates ZO-1 expression by binding ZO-1 promoter in human colon epithelial cells.86 Notably, decreased GATA6 expression was also
seen in the intestinal epithelium of patients with inflammatory bowel diseases.86 These observations indicate a role for Gata6 in the function of the intestinal epithelial barrier and
immune-inflammatory responses, and loss of GATA6 may contribute to the development of NEC by altering the expression of tight junction proteins-coding gene. Given the genetic heterogeneity
of NEC, candidate pathway-targeted or unbiased genomic sequencing approaches are needed to identify novel candidate genes for NEC. Our findings provide novel insights into understanding the
genetic basis underlying NEC associated with CHD and expand the phenotypic spectrum associated with _GATA6_ variants to intestinal abnormalities such as NEC and intestinal perforation. This
study has several limitations that should be noted. First, the in vitro functional assays of the _GATA6_ p.Cys468Tyr variant did not investigate the mechanistic role of the _GATA6_ variant
by evaluation of specific GATA6 target genes or interacting proteins that are specific for cardiac development and CHD. Although pathogenic variation in _GATA6_ is a well-accepted genetic
etiology of CHD and the altered cysteine is located in a highly conserved zinc finger, these additional experimental studies would strengthen our findings. Second, the functional studies
were in vitro assays, therefore, the findings may not translate to the developing human heart, intestine and pancreas. Additional in vivo animal studies are required to confirm the role of
_GATA6_ and this novel _GATA6_ variant on cardiac morphogenesis, as well as intestinal and pancreas development. Lastly, immunofluorescence analysis of both GATA6 wildtype and p.Cys468Tyr
demonstrated an abnormal nuclear localization pattern, however, further analyses are warranted to determine the nature of the protein aggregates and examination of protein expression in
cellular compartments by Western blot. In conclusion, we describe a novel heterozygous _GATA6_ variant (p.Cys468Tyr) resulting in a spectrum of CHD and extracardiac abnormalities. The family
members presented PTA, ASD, BAV, and uniquely, NEC with intestinal perforation. Our findings expand the genotypic and phenotypic spectrum for likely pathogenic variation in _GATA6_,
highlighting gut abnormalities. The combination of early neonatal intestinal perforation and CHD should lead clinical providers to consider the possibility of _GATA6_ pathogenic variation,
which could have clinical implications in the intensive care unit as well as prediction of associated phenotypes and genetic counseling. DATA AVAILABILITY Data presented in current
publication have been deposited in and are available in dbGaP database under dbGaP accession phs002010.v1.p1. Some restrictions apply for dbGaP, as data is available to researchers who meet
access criteria. Data access is through the dbGaP website and researchers apply for data access. Supplied accession number should be used to search the dataset on the website:
https://www.ncbi.nlm.nih.gov/gap/. REFERENCES * Tremblay, M., Sanchez-Ferras, O. & Bouchard, M. Gata transcription factors in development and disease. _Development_ 145, dev164384
(2018). Article PubMed Google Scholar * Peterkin, T., Gibson, A., Loose, M. & Patient, R. The roles of Gata-4, -5 and -6 in vertebrate heart development. _Semin Cell Dev. Biol._ 16,
83–94 (2005). Article CAS PubMed Google Scholar * Afouda, B. A. Towards understanding the gene-specific roles of gata factors in heart development: Does Gata4 lead the way? _Int J. Mol.
Sci._ 23, 5255 (2022). Article CAS PubMed PubMed Central Google Scholar * Pierpont, M. E. et al. Genetic basis for congenital heart disease: Revisited: A Scientific Statement from the
American Heart Association. _Circulation_ 138, e653–e711 (2018). Article PubMed PubMed Central Google Scholar * Yasuhara, J. & Garg, V. Genetics of congenital heart disease: A
narrative review of recent advances and clinical implications. _Transl. Pediatr._ 10, 2366–2386 (2021). Article PubMed PubMed Central Google Scholar * Garg, V. et al. Gata4 mutations
cause human congenital heart defects and reveal an interaction with Tbx5. _Nature_ 424, 443–447 (2003). Article CAS PubMed Google Scholar * Tomita-Mitchell, A., Maslen, C. L., Morris, C.
D., Garg, V. & Goldmuntz, E. Gata4 sequence variants in patients with congenital heart disease. _J. Med Genet_ 44, 779–783 (2007). Article CAS PubMed PubMed Central Google Scholar
* LaHaye, S. et al. Utilization of whole exome sequencing to identify causative mutations in familial congenital heart disease. _Circ. Cardiovasc Genet_ 9, 320–329 (2016). Article CAS
PubMed PubMed Central Google Scholar * Yang, B. et al. Protein-altering and regulatory genetic variants near gata4 implicated in bicuspid aortic valve. _Nat. Commun._ 8, 15481 (2017).
Article CAS PubMed PubMed Central Google Scholar * Dixit, R. et al. Functionally significant, novel gata4 variants are frequently associated with tetralogy of fallot. _Hum. Mutat._ 39,
1957–1972 (2018). Article CAS PubMed Google Scholar * Musfee, F. I. et al. Rare deleterious variants of Notch1, Gata4, Smad6, and Robo4 are enriched in bav with early onset complications
but not in bav with heritable thoracic aortic disease. _Mol. Genet Genom. Med._ 8, e1406 (2020). Article CAS Google Scholar * Padang, R., Bagnall, R. D., Richmond, D. R., Bannon, P. G.
& Semsarian, C. Rare non-synonymous variations in the transcriptional activation domains of Gata5 in bicuspid aortic valve disease. _J. Mol. Cell Cardiol._ 53, 277–281 (2012). Article
CAS PubMed Google Scholar * Wei, D. et al. Gata5 loss-of-function mutations underlie tetralogy of fallot. _Int J. Med. Sci._ 10, 34–42 (2013). Article CAS PubMed Google Scholar *
Jiang, J. Q. et al. Prevalence and spectrum of Gata5 mutations associated with congenital heart disease. _Int J. Cardiol._ 165, 570–573 (2013). Article PubMed Google Scholar * Bonachea,
E. M. et al. Rare Gata5 sequence variants identified in individuals with bicuspid aortic valve. _Pediatr. Res._ 76, 211–216 (2014). Article CAS PubMed PubMed Central Google Scholar *
Shi, L. M. et al. Gata5 loss-of-function mutations associated with congenital bicuspid aortic valve. _Int J. Mol. Med._ 33, 1219–1226 (2014). Article CAS PubMed Google Scholar * Xin, M.
et al. A threshold of Gata4 and Gata6 expression is required for cardiovascular development. _Proc. Natl. Acad. Sci. USA_ 103, 11189–11194 (2006). Article CAS PubMed PubMed Central
Google Scholar * Nemer, G. & Nemer, M. Transcriptional activation of Bmp-4 and regulation of Mammalian organogenesis by Gata-4 and -6. _Dev. Biol._ 254, 131–148 (2003). Article CAS
PubMed Google Scholar * Molkentin, J. D. The zinc finger-containing transcription factors Gata-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. _J.
Biol. Chem._ 275, 38949–38952 (2000). Article CAS PubMed Google Scholar * Lepore, J. J. et al. Gata-6 regulates semaphorin 3c and is required in cardiac neural crest for cardiovascular
morphogenesis. _J. Clin. Invest_ 116, 929–939 (2006). Article CAS PubMed PubMed Central Google Scholar * Kodo, K. et al. Gata6 mutations cause human cardiac outflow tract defects by
disrupting semaphorin-plexin signaling. _Proc. Natl. Acad. Sci. USA_ 106, 13933–13938 (2009). Article CAS PubMed PubMed Central Google Scholar * Lin, X. et al. A novel Gata6 mutation in
patients with tetralogy of fallot or atrial septal defect. _J. Hum. Genet_ 55, 662–667 (2010). Article CAS PubMed Google Scholar * Maitra, M., Koenig, S. N., Srivastava, D. & Garg,
V. Identification of Gata6 sequence variants in patients with congenital heart defects. _Pediatr. Res._ 68, 281–285 (2010). Article CAS PubMed PubMed Central Google Scholar * Kodo, K.
et al. Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. _Circ. J._ 76, 1703–1711 (2012). Article CAS PubMed Google
Scholar * Wang, J. et al. Novel Gata6 mutations associated with congenital ventricular septal defect or tetralogy of fallot. _DNA Cell Biol._ 31, 1610–1617 (2012). Article CAS PubMed
PubMed Central Google Scholar * Huang, R. T., Xue, S., Xu, Y. J. & Yang, Y. Q. Somatic mutations in the Gata6 gene underlie sporadic tetralogy of fallot. _Int J. Mol. Med._ 31, 51–58
(2013). Article CAS PubMed Google Scholar * Zhang, E. et al. Targeted sequencing identifies novel Gata6 variants in a large cohort of patients with conotruncal heart defects. _Gene_ 641,
341–348 (2018). Article CAS PubMed Google Scholar * Škorić-Milosavljević, D. et al. Gata6 mutations: Characterization of two novel patients and a comprehensive overview of the gata6
genotypic and phenotypic spectrum. _Am. J. Med Genet A_ 179, 1836–1845 (2019). Article PubMed PubMed Central Google Scholar * Gharibeh, L. et al. Gata6 regulates aortic valve remodeling,
and its haploinsufficiency leads to right-left type bicuspid aortic valve. _Circulation_ 138, 1025–1038 (2018). Article CAS PubMed PubMed Central Google Scholar * Williams, S. G.,
Byrne, D. J. F. & Keavney, B. D. Rare Gata6 variants associated with risk of congenital heart disease phenotypes in 200,000 Uk biobank exomes. _J. Hum. Genet_ 67, 123–125 (2022). Article
PubMed Google Scholar * Allen, H. L. et al. Gata6 haploinsufficiency causes pancreatic agenesis in humans. _Nat. Genet_ 44, 20–22 (2011). Article PubMed PubMed Central Google Scholar
* Chao, C. S. et al. Novel Gata6 mutations in patients with pancreatic agenesis and congenital heart malformations. _PLoS One_ 10, e0118449 (2015). Article PubMed PubMed Central Google
Scholar * Du, Y. T., Moore, L., Poplawski, N. K. & De Sousa, S. M. C. Familial Gata6 mutation causing variably expressed diabetes mellitus and cardiac and renal abnormalities.
_Endocrinol. Diabetes Metab. Case Rep._ 2019, 19–0022 (2019). PubMed PubMed Central Google Scholar * Sanchez-Lechuga, B. et al. Case report: Adult onset diabetes with partial pancreatic
agenesis and congenital heart disease due to a de novo Gata6 mutation. _BMC Med Genet_ 21, 70 (2020). Article CAS PubMed PubMed Central Google Scholar * Raghuram, N., Marwaha, A.,
Greer, M. C., Gauda, E. & Chitayat, D. Congenital hypothyroidism, cardiac defects, and pancreatic agenesis in an infant with Gata6 mutation. _Am. J. Med Genet A_ 182, 1496–1499 (2020).
Article CAS PubMed Google Scholar * De Franco, E. et al. Gata6 mutations cause a broad phenotypic spectrum of diabetes from pancreatic agenesis to adult-onset diabetes without exocrine
insufficiency. _Diabetes_ 62, 993–997 (2013). Article PubMed PubMed Central Google Scholar * Yu, L. et al. Whole exome sequencing identifies de novo mutations in Gata6 associated with
congenital diaphragmatic hernia. _J. Med Genet_ 51, 197–202 (2014). Article CAS PubMed Google Scholar * Gordon, D. M. et al. Exome sequencing in multiplex families with left-sided
cardiac defects has high yield for disease gene discovery. _PLoS Genet_ 18, e1010236 (2022). Article CAS PubMed PubMed Central Google Scholar * Manivannan, S. N. et al. Novel frameshift
variant in myl2 reveals molecular differences between dominant and recessive forms of hypertrophic cardiomyopathy. _PLoS Genet_ 16, e1008639 (2020). Article CAS PubMed PubMed Central
Google Scholar * Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A Joint Consensus Recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. _Genet Med._ 17, 405–424 (2015). Article PubMed PubMed Central Google Scholar * Ittisoponpisan, S. et al. Can predicted protein 3d
structures provide reliable insights into whether missense variants are disease associated? _J. Mol. Biol._ 431, 2197–2212 (2019). Article CAS PubMed PubMed Central Google Scholar *
Rodrigues, C. H. M., Pires, D. E. V. & Ascher, D. B. Dynamut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. _Protein Sci._ 30, 60–69
(2021). Article CAS PubMed Google Scholar * Edelheit, O., Hanukoglu, A. & Hanukoglu, I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel
to generate mutants for protein structure-function studies. _BMC Biotechnol._ 9, 61 (2009). Article PubMed PubMed Central Google Scholar * Klickstein, J. A., Mukkavalli, S. & Raman,
M. Aggrecount: An unbiased image analysis tool for identifying and quantifying cellular aggregates in a spatially defined manner. _J. Biol. Chem._ 295, 17672–17683 (2020). Article CAS
PubMed PubMed Central Google Scholar * Collett, R. W. & Edwards, J. E. Persistent truncus arteriosus; a classification according to anatomic types. _Surg. Clin. North Am._ 29,
1245–1270 (1949). Article CAS PubMed Google Scholar * Kelly, B. J. et al. Churchill: An ultra-fast, deterministic, highly scalable and balanced parallelization strategy for the discovery
of human genetic variation in clinical and population-scale genomics. _Genome Biol._ 16, 6 (2015). Article PubMed PubMed Central Google Scholar * Ramensky, V., Bork, P. & Sunyaev,
S. Human non-synonymous Snps: Server and survey. _Nucleic Acids Res._ 30, 3894–3900 (2002). Article CAS PubMed PubMed Central Google Scholar * Rentzsch, P., Witten, D., Cooper, G. M.,
Shendure, J. & Kircher, M. Cadd: Predicting the deleteriousness of variants throughout the human genome. _Nucleic Acids Res._ 47, D886–d894 (2019). Article CAS PubMed Google Scholar
* Garber, M. et al. Identifying novel constrained elements by exploiting biased substitution patterns. _Bioinformatics_ 25, i54–i62 (2009). Article CAS PubMed PubMed Central Google
Scholar * Shihab, H. A. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. _Hum. Mutat._ 34, 57–65 (2013).
Article CAS PubMed Google Scholar * Siepel, A. & Haussler, D. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. _Mol. Biol. Evol._ 21, 468–488
(2004). Article CAS PubMed Google Scholar * Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using Gerp++. _PLoS Comput Biol._ 6,
e1001025 (2010). Article PubMed PubMed Central Google Scholar * Ioannidis, N. M. et al. Revel: An ensemble method for predicting the pathogenicity of rare missense variants. _Am. J. Hum.
Genet_ 99, 877–885 (2016). Article CAS PubMed PubMed Central Google Scholar * Quang, D., Chen, Y. & Xie, X. Dann: A deep learning approach for annotating the pathogenicity of
genetic variants. _Bioinformatics_ 31, 761–763 (2015). Article CAS PubMed Google Scholar * Yang, A. et al. Chdgene: A curated database for congenital heart disease genes. _Circ. Genom.
Precis Med_ 15, e003539 (2022). Article CAS PubMed Google Scholar * Zheng, G. F. et al. A novel Gata6 mutation associated with congenital ventricular septal defect. _Int J. Mol. Med._
29, 1065–1071 (2012). CAS PubMed Google Scholar * Homsy, J. et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. _Science_ 350,
1262–1266 (2015). Article CAS PubMed PubMed Central Google Scholar * Jin, S. C. et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands.
_Nat. Genet_ 49, 1593–1601 (2017). Article CAS PubMed PubMed Central Google Scholar * Sharma, A. et al. Gata6 mutations in hipscs inform mechanisms for maldevelopment of the heart,
pancreas, and diaphragm. _Elife_ 9, e53278 (2020). Article CAS PubMed PubMed Central Google Scholar * Trainor, C. D., Ghirlando, R. & Simpson, M. A. Gata Zinc finger interactions
modulate DNA binding and transactivation. _J. Biol. Chem._ 275, 28157–28166 (2000). Article CAS PubMed Google Scholar * Bates, D. L., Chen, Y., Kim, G., Guo, L. & Chen, L. Crystal
structures of multiple gata zinc fingers bound to DNA reveal new insights into DNA recognition and self-association by gata. _J. Mol. Biol._ 381, 1292–1306 (2008). Article CAS PubMed
PubMed Central Google Scholar * Yau, D. et al. Case report: Maternal mosaicism resulting in inheritance of a novel gata6 mutation causing pancreatic agenesis and neonatal diabetes
mellitus. _Diagn. Pathol._ 12, 1 (2017). Article PubMed PubMed Central Google Scholar * Slesnick, T. C., Sachdeva, R., Kreeger, J. R., Pernetz, M. A. & Border, W. L. in
_Echocardiography in Pediatric and Congenital Heart Disease_ 492–507 (2021). * Patel, R. M. & Denning, P. W. Intestinal microbiota and its relationship with necrotizing enterocolitis.
_Pediatr. Res_ 78, 232–238 (2015). Article CAS PubMed PubMed Central Google Scholar * Guthrie, S. O. et al. Necrotizing enterocolitis among neonates in the United States. _J.
Perinatol._ 23, 278–285 (2003). Article PubMed Google Scholar * McElhinney, D. B. et al. Necrotizing enterocolitis in neonates with congenital heart disease: Risk factors and outcomes.
_Pediatrics_ 106, 1080–1087 (2000). Article CAS PubMed Google Scholar * Spinner, J. A. et al. Necrotizing enterocolitis and associated mortality in neonates with congenital heart
disease: A Multi-Institutional Study. _Pediatr. Crit. Care Med_ 21, 228–234 (2020). Article PubMed Google Scholar * Petrosyan, M., Guner, Y. S., Williams, M., Grishin, A. & Ford, H.
R. Current concepts regarding the pathogenesis of necrotizing enterocolitis. _Pediatr. Surg. Int_ 25, 309–318 (2009). Article PubMed Google Scholar * Kodo, K. et al. Regulation of Sema3c
and the interaction between cardiac neural crest and second heart field during outflow tract development. _Sci. Rep._ 7, 6771 (2017). Article PubMed PubMed Central Google Scholar *
Jiang, X. et al. Variants in a cis-regulatory element of Tbx1 in conotruncal heart defect patients impair Gata6-mediated transactivation. _Orphanet J. Rare Dis._ 16, 334 (2021). Article
PubMed PubMed Central Google Scholar * Fenix, A. M. et al. Gain-of-function cardiomyopathic mutations in Rbm20 rewire splicing regulation and re-distribute ribonucleoprotein granules
within processing bodies. _Nat. Commun._ 12, 6324 (2021). Article CAS PubMed PubMed Central Google Scholar * Liu, J. et al. Impairment of the ubiquitin-proteasome system in desminopathy
mouse hearts. _FASEB J._ 20, 362–364 (2006). Article CAS PubMed Google Scholar * Rajasekaran, N. S. et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein
aggregation cardiomyopathy in mice. _Cell_ 130, 427–439 (2007). Article CAS PubMed PubMed Central Google Scholar * Su, H. et al. Cop9 signalosome controls the degradation of cytosolic
misfolded proteins and protects against cardiac proteotoxicity. _Circ. Res_ 117, 956–966 (2015). Article CAS PubMed PubMed Central Google Scholar * Rauch, R. et al. Comprehensive
genotype-phenotype analysis in 230 patients with tetralogy of fallot. _J. Med Genet_ 47, 321–331 (2010). Article CAS PubMed Google Scholar * Walker, E. M., Thompson, C. A. & Battle,
M. A. Gata4 and Gata6 regulate intestinal epithelial cytodifferentiation during development. _Dev. Biol._ 392, 283–294 (2014). Article CAS PubMed PubMed Central Google Scholar * Walker,
E. M., Thompson, C. A., Kohlnhofer, B. M., Faber, M. L. & Battle, M. A. Characterization of the developing small intestine in the absence of either Gata4 or Gata6. _BMC Res Notes_ 7,
902 (2014). Article PubMed PubMed Central Google Scholar * Aronson, B. E., Stapleton, K. A. & Krasinski, S. D. Role of gata factors in development, differentiation, and homeostasis
of the small intestinal epithelium. _Am. J. Physiol. Gastrointest. Liver Physiol._ 306, G474–G490 (2014). Article CAS PubMed PubMed Central Google Scholar * McMillan, T., Girgis, R.
& Sellers, E. A. Neonatal diabetes and protein losing enteropathy: A case report. _BMC Med. Genet_ 17, 32 (2016). Article PubMed PubMed Central Google Scholar * Kim, J. H., Sampath,
V. & Canvasser, J. Challenges in diagnosing necrotizing enterocolitis. _Pediatr. Res._ 88, 16–20 (2020). Article PubMed Google Scholar * Sampath, V. et al. Sigirr genetic variants in
premature infants with necrotizing enterocolitis. _Pediatrics_ 135, e1530–e1534 (2015). Article PubMed PubMed Central Google Scholar * Härtel, C. et al. Nod2 loss-of-function mutations
and risks of necrotizing enterocolitis or focal intestinal perforation in very low-birth-weight infants. _Inflamm. Bowel Dis._ 22, 249–256 (2016). Article PubMed Google Scholar * Cuna,
A., George, L. & Sampath, V. Genetic predisposition to necrotizing enterocolitis in premature infants: current knowledge, challenges, and future directions. _Semin Fetal Neonatal Med._
23, 387–393 (2018). Article PubMed PubMed Central Google Scholar * Bein, A., Eventov-Friedman, S., Arbell, D. & Schwartz, B. Intestinal tight junctions are severely altered in nec
preterm neonates. _Pediatr. Neonatol._ 59, 464–473 (2018). Article PubMed Google Scholar * Högberg, N., Stenbäck, A., Carlsson, P. O., Wanders, A. & Lilja, H. E. Genes regulating
tight junctions and cell adhesion are altered in early experimental necrotizing enterocolitis. _J. Pediatr. Surg._ 48, 2308–2312 (2013). Article PubMed Google Scholar * Laudisi, F. et al.
Gata6 deficiency leads to epithelial barrier dysfunction and enhances susceptibility to gut inflammation. _J. Crohns Colitis_ 16, 301–311 (2022). Article PubMed Google Scholar Download
references ACKNOWLEDGEMENTS We thank the family members for study participation, Gloria Zender in Cell Line Core and Samantha Fichtner and Jade Hayden in Heart Center Clinical Research Core.
FUNDING This work was supported in part by funding from National Institutes of Health (R01 HL109758) to V.G., K.L.M, and P.W. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Center for
Cardiovascular Research, Abigail Wexner Research Institute, Nationwide Children’s Hospital, Columbus, OH, USA Jun Yasuhara, Sathiya N. Manivannan, Uddalak Majumdar, Mona Aljuhani, Katherine
Myers, Kim L. McBride & Vidu Garg * The Heart Center, Nationwide Children’s Hospital, Columbus, OH, USA Jun Yasuhara, Sathiya N. Manivannan, Uddalak Majumdar, Mona Aljuhani, Katherine
Myers, Corey Stiver, Mark Galantowicz, Kim L. McBride & Vidu Garg * Institute for Genomic Medicine, Nationwide Children’s Hospital, Columbus, OH, USA David M. Gordon, Patrick J. Lawrence
& Peter White * Department of Pediatrics, The Ohio State University, Columbus, OH, USA Corey Stiver, Amee M. Bigelow, Kim L. McBride, Peter White & Vidu Garg * Division of Pediatric
Cardiology, Department of Pediatrics, Keio University School of Medicine, Tokyo, Japan Hiroyuki Yamagishi * Division of Genetic and Genomic Medicine, Nationwide Children’s Hospital,
Columbus, OH, USA Kim L. McBride * Department of Molecular Genetics, The Ohio State University, Columbus, OH, USA Vidu Garg Authors * Jun Yasuhara View author publications You can also
search for this author inPubMed Google Scholar * Sathiya N. Manivannan View author publications You can also search for this author inPubMed Google Scholar * Uddalak Majumdar View author
publications You can also search for this author inPubMed Google Scholar * David M. Gordon View author publications You can also search for this author inPubMed Google Scholar * Patrick J.
Lawrence View author publications You can also search for this author inPubMed Google Scholar * Mona Aljuhani View author publications You can also search for this author inPubMed Google
Scholar * Katherine Myers View author publications You can also search for this author inPubMed Google Scholar * Corey Stiver View author publications You can also search for this author
inPubMed Google Scholar * Amee M. Bigelow View author publications You can also search for this author inPubMed Google Scholar * Mark Galantowicz View author publications You can also search
for this author inPubMed Google Scholar * Hiroyuki Yamagishi View author publications You can also search for this author inPubMed Google Scholar * Kim L. McBride View author publications
You can also search for this author inPubMed Google Scholar * Peter White View author publications You can also search for this author inPubMed Google Scholar * Vidu Garg View author
publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS J.Y. and V.G. conceived and designed the study. K.M, C.S., K.L.M, and V.G. enrolled patients. J.Y.,
M.A., K.M., and C.S. collected clinical data. J.Y., S.N.M., and U.M. performed and acquired experimental data. J.Y., S.N.M., D.M.G., P.J.L, and P.W. performed bioinformatics analyses. J.Y.,
S.N.M., U.M., D.M.G., P.J.L, K.L.M., P.W., and V.G. analyzed and interpreted the data. J.Y. and V.G. drafted the manuscript. C.S., A.M.B., M.G., H.Y., K.L.M., P.W., and V.G. critically
reviewed the manuscript. All authors read and approved the final version of the manuscript for publication. CORRESPONDING AUTHOR Correspondence to Vidu Garg. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. INFORMED CONSENT Statement of written consent was obtained as described in the Ethics statement. ADDITIONAL INFORMATION PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY MATERIAL RIGHTS AND
PERMISSIONS Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s);
author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law. Reprints and permissions ABOUT THIS
ARTICLE CITE THIS ARTICLE Yasuhara, J., Manivannan, S.N., Majumdar, U. _et al._ Novel pathogenic _GATA6_ variant associated with congenital heart disease, diabetes mellitus and necrotizing
enterocolitis. _Pediatr Res_ 95, 146–155 (2024). https://doi.org/10.1038/s41390-023-02811-y Download citation * Received: 27 February 2023 * Revised: 11 August 2023 * Accepted: 21 August
2023 * Published: 12 September 2023 * Issue Date: January 2024 * DOI: https://doi.org/10.1038/s41390-023-02811-y SHARE THIS ARTICLE Anyone you share the following link with will be able to
read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing
initiative
Trending News
Nature chemical biology - volume 13 issue 8, august 2017The cover depicts conidiophores of the fungus _Aspergillus nidulans_ carrying a fungal artificial chromosome (FAC), imag...
Optical refrigeration | Nature PhotonicsABSTRACT The idea of cooling a solid-state optical material by simply shining a laser beam onto it may seem counterintui...
Nature chemical biology - volume 13 issue 5, may 2017A new autoinducer–receptor pair, 3,5-dimethylpyrazin-2-ol (DPO)–VqmA, acts in parallel to canonical _Vibrio cholerae_ qu...
Page Not Found (404) | WFAE 90.7 - Charlotte's NPR News SourceWe're sorry; the page you're looking for cannot be found. Please use the search option at the top of the page to find wh...
6. Epigenomic changes in human disease and during cancer progressionEpigenome profiling reveals multi-tissue regulatory dynamics associated with progression of cancer, Alzheimer’s disease ...
Latests News
Novel pathogenic gata6 variant associated with congenital heart disease, diabetes mellitus and necrotizing enterocolitisABSTRACT BACKGROUND Pathogenic _GATA6_ variants have been associated with congenital heart disease (CHD) and a spectrum ...
Vaccination with helper-dependent adenovirus enhances the generation of transgene-specific ctlABSTRACT Recombinant adenoviral vectors (AdV) have been used experimentally as vaccines to present antigenic transgenes ...
Exchange on nations and identities | thearticleFirst {{register.errors.names}} Last Gender What's this for? Age bracket What's this for? This is to help us s...
Nature reviews cardiology - volume 18 issue 2, february 2021Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best expe...
Guided Imagery: The BeachMemorial Day Sale! Join AARP for just $11 per year with a 5-year membership Join now and get a FREE gift. Expires 6/4 G...