Gastric cancer: genome damaged by bugs
Gastric cancer: genome damaged by bugs"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Gastric cancer (GC) is one of the leading causes of cancer-related death worldwide. The role of the microorganisms in gastric tumorigenesis attracts much attention in recent years.
These microorganisms include bacteria, virus, and fungi. Among them, _Helicobacter pylori_ (_H. pylori_) infection is by far the most important risk factor for GC development, with special
reference to the early-onset cases. _H. pylori_ targets multiple cellular components by utilizing various virulence factors to modulate the host proliferation, apoptosis, migration, and
inflammatory response. Epstein–Barr virus (EBV) serves as another major risk factor in gastric carcinogenesis. The virus protein, EBER noncoding RNA, and EBV miRNAs contribute to the
tumorigenesis by modulating host genome methylation and gene expression. In this review, we summarized the related reports about the colonized microorganism in the stomach and discussed
their specific roles in gastric tumorigenesis. Meanwhile, we highlighted the therapeutic significance of eradicating the microorganisms in GC treatment. SIMILAR CONTENT BEING VIEWED BY
OTHERS _HELICOBACTER PYLORI_, MICROBIOTA AND GASTRIC CANCER — PRINCIPLES OF MICROORGANISM-DRIVEN CARCINOGENESIS Article 26 February 2025 COMPARABLE GENETIC ALTERATION PROFILES BETWEEN
GASTRIC CANCERS WITH CURRENT AND PAST _HELICOBACTER PYLORI_ INFECTION Article Open access 06 December 2021 THE NOVEL ZEB1-UPREGULATED PROTEIN PRTG INDUCED BY _HELICOBACTER PYLORI_ INFECTION
PROMOTES GASTRIC CARCINOGENESIS THROUGH THE CGMP/PKG SIGNALING PATHWAY Article Open access 04 February 2021 INTRODUCTION Gastric cancer (GC) is the second leading cause of cancer-related
death in the world [1]. GC mainly occurs in Asia, Latin America, and Central and Eastern Europe, however, it is no longer a common disease in North America and part of Western Europe [2]. GC
can be separated into two types according to the locus, gastric adenocarcinomas and gastro-esophageal-junction adenocarcinomas [3]. Gastric adenocarcinoma can also be subdivided
histologically into intestinal and diffuse types by Lauren’s classification. In 2014, The Cancer Genome Atlas (TCGA) research network has described a comprehensive molecular evaluation on
295 primary gastric adenocarcinomas. They proposed a molecular classification dividing GC into four subtypes: positive for Epstein–Barr virus (EBV) (9%), microsatellite unstable tumors
(22%), genomically stable tumors (20%), and chromosomally unstable tumors (50%) [4]. In 2015, the Asian Cancer Research Group (ACRG) proposed another molecular classification associated with
clinical outcome and defined GC as four distinct molecular subtypes: microsatellite instability (MSI), microsatellite stable with epithelial-to-mesenchymal transition features (MSS/EMT),
MSS/TP53 mutant (MSS/TP53+), and MSS/TP53 wild type (MSS/TP53–) [5]. Identification of these subtypes sheds new lights on the prognosis and clinical treatment [6]. More than 15% of the tumor
cases were attributed to infectious pathogens. The proportion was even higher in less developed countries or regions (22.9%) [7]. The infectious pathogens include viruses, bacteria, and
parasites. Among the pathogens, _Helicobacter pylori_ (_H. pylori_), human papillomavirus, hepatitis B virus (HBV), and hepatitis C virus together attributed to 2 million new cancer cases
worldwide in 2012. They induced the tumorigenicity of the stomach, liver, and cervix. Of note, HBV and _H. pylori_ have most vicious contributions to the tumor burden in China [8]. _H.
pylori_ and EBV are the most well-known pathogens in gastric carcinogenesis. _H. pylori_ is an important risk factor found in 65–80% of primary GCs, while EBV leads to 10% of the GC cases.
Besides, it has been reported other microorganisms are also associated with gastric malignancies. Accompanied with the development of strategies for manipulating infectious agents,
opportunities are emerging to prevent and cure the infection-related cancers. Here, we comprehensively reviewed the role of microbiome in promoting gastric carcinogenesis. BACTERIOME IN
GASTRIC CARCINOGENESIS Because of the acid production, stomach was thought as a sterile organ previously. However, in recent years, culture independent methods have been developed to
facilitate the identification of various bacteria species in human stomach. It is believed that apart from the predominant bacteria _H. pylori_, multiple kinds of bacteria were coexisting in
human stomach, although little is known about their associations with GC progression. INFECTION OF _H. PYLORI_ _H. pylori_ infection is the most popular chronical bacterial infection
worldwide. More than 50% of the world population are infected with _H. pylori_, however, over 80% of infections are asymptomatic [9]. The transmission of _H. pylori_ is implicated with
fecal/oral, oral/oral, or gastric/oral pathways [10]. Part of the infections develop coexisting gastritis for several years, and the persistent infection might develop into gastric atrophy
followed by intestinal metaplasia, dysplasia, and eventually adenocarcinoma [6]. World Health Organization designates _H. pylori_ as a class I carcinogen because of its chronic infection as
the strongest risk factor for gastric adenocarcinoma. It was estimated that 90% of all noncardia GCs are associated with _H. pylori_ [11]. A study with 1526 Japanese population found the
increasing risk of GC development in patients infected with _H. pylori_ compared with the uninfected ones [12]. The eradication of _H. pylori_ significantly decreased the occurrence of GC,
suggesting that _H. pylori_ might influence early stages in gastric carcinogenesis [13]. MOLECULAR PATHOGENESIS OF _H. PYLORI_-RELATED GC Environmental factors have long been considered to
play dispensable roles in GC. High salt intake was found significantly associated with GC especially in the context of _H. pylori_ infection and atrophic gastritis [14]. It was also believed
that the risk of GC increased in the subjects with both smoking habit and _H. pylori_ infection [15]. It has been puzzling about _H. pylori_ infection, although half of the population
infected with _H. pylori_ worldwide, only a minority of colonized individuals (1–2%) develops tumors. The low morbidity indicates the impact of different strains in tumor initiation and
development. Different strains of _H. pylori_ play diverse roles in driving GC. _H. pylori_ can be subdivided into bacterial oncoprotein cytotoxin-associated gene A (CagA) positive and CagA
negative strains. In a meta-analysis, patients infected with CagA positive strains demonstrate a higher risk of GC [16], which was consistent with previous reports that individuals with CagA
antibodies have a higher risk of tumor [17,18,19,20]. Transgenic mice bearing CagA appears gastric neoplasms development, confirming that CagA is a bacteric oncoprotein [21]. However, the
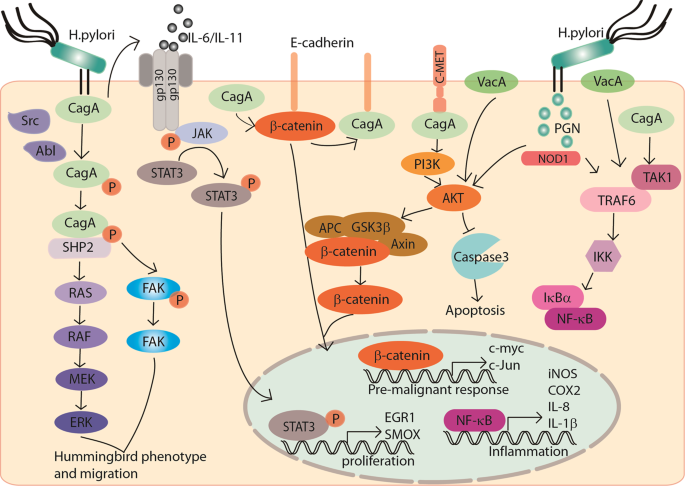
mechanism seems particularly complex. _H. pylori_ injects CagA into the host gastric epithelial cells with the activation of integrin [22]. Moreover, CagA undergoes tyrosine phosphorylation
by Src family kinases or Abl kinase and subsequently activates multiple signaling pathways. For instance, phosphorylated CagA interacts with activated SHP2. CagA–SHP2 potentiates the
magnitude of Erk-MAP kinase signaling in both Ras-dependent and Ras-independent manners [23]. CagA–SHP2 also dephosphorylates focal adhesion kinase (FAK) and mediates cell–extracellular
matrix interaction. Both signaling lead to a cellular morphological change, which is called hummingbird phenotype, thus to increase the cell migration abilities [24]. In addition,
nonphosphorylated CagA impairs intracellular signaling networks. The nonphosphorylated intracellular CagA interacts with E-cadherin to disrupt the E-cadherin–β-catenin complex. It thus
induces nuclear β-catenin accumulation, allowing transcription of the target genes associated with carcinogenesis. Meanwhile, CagA was reported to directly activate β-catenin by interacting
with MET and activating PI3K–AKT signaling [25, 26]. CagA activates the signal transducer and activator of transcription 3 (STAT3) pathway. The activated STAT3 pathway is driven by the host
immune response and is associated with _H. pylori_-induced gastritis and cancer progression, independent of CagA phosphorylation [27,28,29]. In a recent study, CagA also binds to 25 known
factors in the host cells to hijack various signaling pathways related to inflammation, proliferation, genetic instability, cell polarity, and apoptosis [30]. Apart from CagA, the Cag
secretion system also delivers _H. pylori_ peptidoglycan into the host cells through outer membrane vesicles. The peptidoglycan subsequently activates PI3K–AKT and regulates cell migration,
proliferation, and apoptosis [31]. Apart from Cag, vacuolating toxin A (VacA) is another major virulence determinant of _H. pylori_. _H. pylori_ gene _vacA_ encodes the secreted protein
VacA. VacA has been reported to link to multiple cellular processes, such as vacuolation, membrane-channel formation, apoptosis, proinflammatory response, and malignancy [32]. Although all
of the _H. pylori_ strains contain _vacA_, there is variation in the _vacA_ structure. Among them, s1m1i1d1 type strains are strongly associated with gastric adenocarcinoma. Nakayama et al.
reported that VacA activates β-catenin through PI3K-dependent manner [33]. Approximately, 4% of the _H. pylori_ genome encodes integral outer membrane proteins (OMPs) [34, 35], which are
subdivided as 5 families [36]. Some of them functioned as adherence factors, such as sialic acid-binding adhesin, blood-group-antigen-binding adhesin, adherence-associated lipoprotein A and
B, outer inflammatory protein A (OipA), and _Helicobacter_ OMP Q. Most of them are linked with poor clinical outcomes. OipA was identified as a proinflammatory response inducing protein and
knockout of this gene can reduce interleukin (IL)-8 production [37]. In patient samples, it was confirmed that OipA was significantly associated with gastric inflammation and IL-8 levels
[38]. Basically, OipA is involved in the attachment of _H. pylori_ to gastric epithelial cells, which is important for the initiation and development of GC. In addition, inactivation of OipA
decreases the incidence of carcinoma by attenuating β-catenin nuclear translocation [39]. The aberrant host genetic changes are also crucial for the interaction of _H. pylori_ and gastric
epithelium cells. Polymorphisms in IL-1β and its endogenous receptor antagonist affect gastric mucosal IL-1β production in response to infection of _H. pylori_ and are associated with GC
occurrence [40,41,42]. In addition, the combination of HLA class II and IL-10–592A/C polymorphisms affect the susceptibility to GC development in _H. pylori_-infected Japanese individuals
[43]. The causal relationship between inflammation and cancer has been well recognized. An individual infected with _H. pylori_ has a bigger chance to develop chronic inflammation. _H.
pylori_ utilizes virulence factors CagA, VacA, and peptidoglycan to upregulate proinflammatory cytokines such as IL-1, IL-6, IL-8, TNF-α, and NF-κB, to activate NF-κB signaling cascade in
gastric epithelial cells and circulating immune cells [44]. The production of cytokine triggers activation and migration of leukocytes, and regulation cascade of cytokines, chemokine, and
adhesions. Granulocyte-macrophage colony-stimulating factor, a growth factor facilitating white cell differentiation, was found in _H. pylori_-infected antral biopsies and human gastric
epithelial cells [45]. Besides, inflammation modulators cyclooxygenase-2, which convers arachidonic acid to prostaglandins to induce inflammatory reactions, was significantly higher in _H.
pylori_-infected gastric epithelia cells [46]. Apart from the cytokine release, lipopolysaccharide (LPS), VacA, and _H. pylori_ neutrophil activating protein contribute to induce reactive
oxygen species (ROS) or reactive nitrogen species (RNS) in gastric epithelial cells and inflammatory cells. The generation of intracellular ROS and RNS are found relating to the pathogenesis
of _H. pylori_-associated GC. In addition, _H. pylori_-induced chronic inflammation leads to aberrant DNA methylation, which is the major cause of _H. pylori_-associated GC. On the other
hand, when _H. pylori_-induced inflammation was suppressed by cyclosporine A in animal model, induction of aberrant DNA methylation was also suppressed [47, 48]. Methylation on
tumor-suppressor genes can inactivate the gene expression and promotes cancer development. For example, promoter methylation in E-cadherin, an epithelial marker, has been detected in _H.
pylori_-infected stomach [49]. Regarding to these studies, _H. pylori_-induced chronic inflammation is essential for both initiation and the development of GC. The potential molecular
network of _H. pylori_ and oncogenic signaling pathways in gastric carcinogenesis are summarized in Fig. 1. HOST IMMUNITY IN _H. PYLORI_-RELATED GC The host immune system is the formidable
barrier to prevent _H. pylori_ infection. The immune system includes innate immune response and adaptive immune response. Innate immune response is the first-line defense. Epithelial cells,
dendritic cells, monocytes, macrophages, and neutrophils could play important roles in defending _H. pylori_ infection. Pathogen-associated molecular patterns of _H. pylori_, such as,
peptidoglycan, LPS, lipoproteins, and flagellins are recognized by pattern recognition receptors (PRRs). Toll-like receptors, C-type lectin receptors, NOD-like receptors, and RIG-like
receptors are members of the PRR family. The engagement of PRR then triggers the activation of multiple signaling cascades that culminate in NF-κB activation and immune effectors production.
Such an immune response could induce a chronic inflammation, which has been shown closely associated with molecular pathogenesis of _H. pylori_-related GC. However, in adaptive immune
response, _H. pylori_ can be recognized and presented by antigen-presenting cells (APCs), such as dendritic cell [50], neutrophil, macrophage, and epithelial cells [51]. The APCs produce
cytokines to stimulate naive CD4+ T cells and induce antigen-specific responses in Th1 cells [52, 53] and Th17 cells [54,55,56]. The Th1 cells and Th17 cells are critical for the control of
_H. pylori_ infection, however they are also associated with increased gastritis as well as GC [54, 57,58,59,60]. At the same times, the T regulatory (Treg) cell response is also observed,
which drives immune tolerance and suppresses Th1- and Th17-mediated immunity against _H. pylori_ infection [61, 62]. It has been reported that B cells and antibodies are not required for
clearing the _H. pylori_, rather, they might be detrimental to elimination of the bacteria [63]. DIAGNOSIS AND TREATMENT OF _H. PYLORI_ _H. pylori_ should be tested in patients with
dyspepsia if the local _H. pylori_ prevalence exceeds 10%. The testing can be performed by noninvasive and invasive methods. The noninvasive methods include the urea breath tests and fecal
antigen test. Serologic test and invasive testing strategies require upper endoscopy, biopsy urease (campylobacter-like organism) test, histologic assessment, and culture [64]. The
eradication of _H. pylori_ dramatically decreases the presence of premalignant lesions and reduce the GC risk in infected individuals. Anti-_H. pylori_ therapy is an effective means for GC
prevention and there are various proposed treatment regimens for _H. pylori_ eradication [65]. Traditional treatment regimens include standard triple therapy (PPI, amoxicillin, and
clarithromycin), bismuth quadruple PBMT therapy (PPI, bismuth, metronidazole, and tetracycline), or a treatment including PPI, clarithromycin, and metronidazole. However, with increasing
clarithromycin resistance, another regimen concomitant nonbismuth therapy PAMC (PPI, amoxicillin, metronidazole, and clarithromycin) was proposed. The first-line treatment was recommended
with a 14-day course of either concomitant PAMC therapy or bismuth quadruple PBMT therapy, according to the 2016 Toronto Consensus guidelines [66]. The 2016 Maastricht V/Florence Consensus
Report recommends first-line treatment with a 14-day course of bismuth quadruple PBMT therapy or concomitant PAMC therapy in high clarithromycin resistance areas (>15% resistance). A
standard triple therapy or bismuth quadruple PBMT therapy in low clarithromycin resistance (<15% resistance) areas is also proposed by this report [67]. OTHER BACTERIA IN GC In 2006, Bik
et al. used a small subunit 16S rDNA clone library approach identified 128 phylotypes belonging to five phyla (Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, and Fusobacteria) in
23 human gastric biopsies [68]. Lately, 133 phylotypes were identified by Li et al. and 59 families were detected by Delgado et al. [69, 70], which were quite similar from both phyla and
genera level. It reflects the significance of the bacterial homeostasis in stomach. Loss of bacterial homeostasis might be a reason in driving GC progression. Coker et al. reported that
microbial composition was changed, and bacterial interactions were different across stages of gastric carcinogenesis, indicating the presence of microbial dysbiosis in gastric
carcinogenesis. They also found potential roles of some microbial such as _Peptostreptococcus stomatis_, _Dialister pneumosintes_, _Slackia_ exigua, _Parvimonas micra_, and _Streptococcus
anginosus_ in GC progression [71]. It was also reported that a consistent increase of lactic acid bacteria promotes GC by a number of mechanisms such as supply of exogenous lactate,
production of ROS, and N-nitroso compounds, as well as anti-_H. pylori_ properties [72]. Notably, _H. pylori_ and other bacteria might affect each other in the stomach, but the causality has
not yet been clearly explained. As currently known, bacteria colonies in the stomach could affect the outcome of _H. pylori_ infection and the progression of GC. On the other side, _H.
pylori_ infection may influence the density of bacteria. In animal model, long-term _H. pylori_ infection affects the bacterial composition of the gastric microbiota. Maldonado-Contreras et
al. reported a higher abundance of Proteobacteria, Spirochetes, and Acidobacteria, and a decreased abundance of Actinobacteria, Bacteroidetes, and Firmicutes in _H. pylori_-positive patients
compared with _H. pylori_-negative subjects [73]. A microbial diversity analysis showed that compared with negative subjects, both of the species and Shannon index were increased in
subjects with past or current _H. pylori_-infected subjects, indicating the alterations of fecal microbiota, especially Bacteroidetes, Firmicutes, and Proteobacteria, may be involved in the
process of _H. pylori_-related gastric lesion progression [74]. However, some reports indicated that chronic _H. pylori_ infection does not alter the microbiota of stomach [68, 71, 75, 76],
suggesting the relationship between _H. pylori_ infection and the gastric microbiota dysbiosis is still controversial [77, 78]. EBV IN GASTRIC CARCINOGENESIS The mammalian virome is
constituted of viruses that infect host cells, virus-derived elements in human chromosomes, and viruses that infect the broad array of other types of organisms [79]. It was reported that
EBV, CMV, and HHV6 can be detected in gastric tumors [80]. Among them, EBV is the most prominent one. THE STRUCTURE OF EBV More than 90% of adults have been infected by EBV [81], and it is
asymptomatic in the majority of carriers. However, some of the infections can cause infectious mononucleosis. EBV is classified as a group I carcinogen by the International Agency for
Research on Cancer, since the latently infection estimated to be responsible for 200,000 cancers cases worldwide [82], such as Burkitt lymphoma, hemophagocytic lymphohistiocytosis, Hodgkin’s
lymphoma, GC, and nasopharyngeal carcinoma (NPC). Until now, approved vaccines for EBV have not been available. However, a vaccine targeting the EBV glycoprotein gp350 has been developed to
reduce the incidence of infectious mononucleosis and the efficacy has been proved [83]. EBV belongs to Herpesviridae containing an ~172 kb liner form dsDNA genome. The expression products
cover 80 proteins and 46 functional small-untranslated RNAs. EBV prefers to infect B cell and epithelial cells. After entry, like all kind of herpesviruses, EBV has two distinct life cycles:
lytic replication and latency. However, upon EBV de novo infection, it takes latency infection firstly. During latency, viral genomes exist as extrachromosomal episomes in the nucleus and
only express some latent proteins (EBV-determined nuclear antigen 1 (EBNA1), 2, 3A, 3B, 3C, and EBNA-LP; latent membrane protein 1 (LMP1) and LMP2), noncoding RNA (EBER1 and EBER2), and
viral miRNAs (BHRF1-miRNA and BART-miRNA) (Fig. 2a). EBV latency is categorized by three latency types (latency I–III), which have different latency protein expression patterns depending on
the type of cell infected. Several different kinds of latency were shown schematically (Fig. 2b). The lytic infection is triggered by several factors from the latent state. Then, nearly 80
proteins are encoded to facilitate the viral particle formation and release into the extracellular space. ESTABLISHMENT OF EBV INFECTION IN STOMACH EPITHELIAL CELLS The first puzzle about
EBV-associated gastric carcinoma (EBVaGC) is how EBV infects gastric epithelial cells, as the EBV infection often occurs in B lymphocytes and the oral epithelium. It is possible that the
EBV-contained saliva is ingested and EBV infects the epithelial cells directly. Another explanation is that EBV is reactivated somehow in B lymphocytes in stomach and released to infect
epithelial cells [84]. Ephrin receptor A2 as well as integrins and nonmuscle myosin heavy chain IIA (NMHCIIA) serve as cofactors and play an important role in EBV epithelial cell entry
[85,86,87,88]. Coculturing of epithelial cells with EBV-positive lymphocyte cells showed about 800 fold higher efficiency of infection than cell-free infection, suggesting the possibility of
direct cell-to-cell mediated virus infection [89]. It was proposed that EBV-infected lymphocytes contacts with epithelial cells via integrin β1/β2, and then promotes cell-to-cell contact by
translocating intracellular adhesion molecule-1 to the cell surface. At last, the viral particle is transmitted by clathrin-mediated endocytosis pathway [90]. After endocytosis, the EBV-DNA
is transported to nucleus, where the naked linear DNA genomes are assembled into a functional circular mini-chromosome. After circulation, viral genome chromatinization can effectively
protect it from DNA damage and offer tight regulation of gene expression [91]. The CpG motifs of viral genome are widely methylated and by this way, latent infection is successfully
established. The infection and latency processes were summarized in Fig. 3. It is well known that EBV LMP1 and nuclear antigen 2 (EBNA2) play major roles in EBV-induced oncogenesis. However
both of them were rarely detected in gastric adenocarcinoma cells [92,93,94]. Instead, EBNA1 expression was confirmed [93, 95]. It was reported that transcription of EBNA was initiated from
EBNA promoters, Qp but not Cp or Wp, which may result in the absent expression of EBNA2 [96]. In addition, BZLF1 is expressed in a proportion of the tumors, suggesting the switch from latent
to lytic infection [92]. THE HISTOPATHOLOGICAL FEATURES OF EBVAGC In 1990, Burke et al. firstly detected EBV in lymphoepithelial carcinoma of the stomach, which was similar to
undifferentiated nasopharyngeal lymphoepithelioma [97]. However, Shibata subsequently found that EBV is involved not only in the rare gastric lymphoepithelioma-like cancers, but also in
gastric adenocarcinomas. They demonstrated that the EBV genomes were specifically present within the gastric carcinoma cells and adjacent dysplastic epithelium but were absent in surrounding
normal cells [98, 99]. The result was confirmed by polymerase chain reaction (PCR) and in situ hybridization (ISH) in variety of studies [92, 94, 100,101,102,103]. Since the EBV-positive
tumor cells were from a single clonal proliferation [93, 102, 104], and EBV was not generally detected in normal stromal cells, metaplasia, gastric mucosa, and lymphocytes [93, 99, 103,
104], EBV infection was believed to occur in the dysplastic phase and related to gastric carcinogenesis. In gastric carcinoma with lymphoid stroma, all cases are EBV-positive tumors.
However, in gastric adenocarcinomas, only a small fraction of the cases shows EBV positive. It is believed that EBV plays distinct roles in etiology of these two types of GC [92, 98].
EBVaGC-associated mortality was estimated to be 70,000 worldwide each year [105]. Epidemiological studies show that male EBV-positive GC patients were twice than female [99, 106] and type 1
strain is most prevalent one in gastric carcinoma [107, 108]. EBVaGC has distinctive clinical characteristics compared with EBV-negative cases. EBVaGC often appears in the upper part of the
stomach and has a diffuse-type histology with lymphoid infiltration [109]. By analyzing individual-level data on 4599 GC patients from 13 studies, it was demonstrated that EBV positivity is
a powerful prognostic indicator of GC. In addition, the report also indicated that patients with EBV-positive GC had a better survival than EBV-negative ones [110], because of the high
degree of homogeneity in EBVaGCs compared with EBV-negative cases. Furthermore, most of the altered genes in EBVaGCs are immune response related genes leading to more immune cells to migrate
into the microenvironment, compared with EBV-negative GC. The recruitment of immune cells contributes to the better clinical outcome for EBVaGC cases [111]. Besides, CD204-positive M2-type
tumor-associated macrophages, which were associated with the aggressive behavior of tumors, exhibit low density in EBVaGCs, partly explaining the favorable outcomes [112]. Recently,
comprehensive molecular characterization of GC presents several distinct molecular features and epigenetic alterations of EBVaGC, including lack of _TP53_ mutations, frequent _PI3K_
mutations, and a high degree of CpG methylation in the tumor cell genome [113]. THE MOLECULAR PATHOGENESIS OF EBVAGC To date, the mechanism of EBVaGC has not yet been comprehensively
deciphered. In general, virologic aspects and genetic abnormalities of host cells co-potentiate the tumor development. As for virologic aspects, since EBV-positive GC is in latency type I,
only EBERs, EBNA1, and miR-BARTs are highly expressed, while LMP2A could be detected in some cases [96, 114]. Meanwhile, genetic abnormalities of host cells caused by EBV infection, such as
aberrant DNA methylation, attract more and more attention these years. The methylation of CpG DNA of the host genome is also caused by the establishment of EBV latent infection and the
expression of the EBV latent genes. PROMOTING ROLES OF VIROLOGIC GENES IN GC PATHOGENESIS EBERs are viral nonpolyadenylated RNA, which is abundantly expressed in latently EBV-infected cells.
Because of their abundance, EBERs serve as the most reliable and sensitive target by ISH to detect EBV infection in tissues. It plays a role in cell proliferation, apoptosis, and antiviral
innate immunity. However, only a few studies investigated the roles of EBERs in EBV-mediated oncogenesis. EBER1 upregulates the expression of insulin growth factor 1, which promotes
proliferation of EBVaGC cells [115]. Another work showed that EBERs induce chemoresistance and enhance cellular migration in coordination with IL-6-STAT3 signaling pathway [116]. EBERs as
well as BARF0, EBNA1, and LMP2A contribute to the downregulation of miR-200 family, resulting in E-cadherin expression reduction, which is a crucial step in the carcinogenesis of EBVaGC
[117]. EBNA1 is an essential molecule for EBV latency infection. It binds to viral oriP sequence in a sequence dependent manner and tethers EBV episomes onto host cell chromosomes, which is
essential for episomal maintenance. EBNA1 also functions as a transactivator of the viral genes. In EBVaGC, EBNA1 enhances tumorigenicity in mouse model [118]. It was also reported to cause
loss of promyelocitic leukemia (PML) nuclear bodies (NBs), resulting in impaired responses to DNA damage and promotion of cell survival [119]. In addition, EBNA1 induces ROS accumulation
mediated by miR-34a and NOX2 to regulate the tumor cell viability [120]. LMP2A was detected in half of the EBVaGC cases [121]. Fukayama et al. found that LMP2A activates the NF-κB-survivin
pathway to rescue EBV-infected epithelial cells from serum deprivation, which may play a role in the progression of EBV-infected GC [122]. By using a recombinant adenoviral expression
vector, Liu et al. found that LMP2A plays an important role in pathogenesis of EBVaGC through regulating cyclin E expression and S phase cell ratio [123]. Besides, LMP2A mediates Notch
signaling to elevate mitochondrial fission and promote cellular migration [124]. In addition, LMP2A could also downregulate HLA to evade the immune response of the malignant cells [125]. It
can activate PI3K/Akt pathway to mediate the transformation process and inhibit TGFβ1-induced apoptosis, which provides a clonal selective advantage for EBV-infected cells during tumor
development [126, 127]. LMP2A upregulates miR-155–5p though NF-κB pathway and this will lead to the inhibition of Smad2 and p-Smad2 [128]. Apart from the direct modulating effects on
tumorigenesis, LMP2A also promotes malignancy by inducing epigenetic modifications of the host genome [129]. Recent studies imply that miR-BARTs contribute to EBV-associated epithelial
carcinogenesis. The miR-BARTs are abundantly expressed in EBV-infected GCs cell line, but not in EBV-transformed lymphocytes [4, 130]. By using EBV-infected AGS cell line (AGS-EBV), the
expression of miR-BARTs was quite rich but the expression of the viral protein was limited [131, 132]. EBV miRNAs contribute to the initiation and development of EBVaGC by targeting multiple
host proteins to mediate cell proliferation, transformation, senescence, apoptosis, and immune response. A comprehensive profiling of EBV miRNAs in EBVaGC was constructed by Tsai et al. and
they found the deletion of miR-BART9 could increase E-cadherin expression and decrease proliferative and invasive ability [133]. BART3–3p plays an important role in inhibiting the
senescence of GC cells by targeting _TP53_ [134]. As for apoptosis, it was reported that BART5–3p directly targets _TP53_, leading to acceleration of the cell cycle progress and inhibition
of cell apoptosis [135]. Besides, EBV encoded miR-BART5 could target p53 upregulated modulator of apoptosis (PUMA), which is a proapoptotic protein belonging to the Bcl-2 family, to
counteract apoptosis and promote cellular survival [136]. In addition to PUMA, it was reported that miR-BART9, 11, and 12 strongly downregulate Bim, which is also a member of Bcl-2 family
[137]. By comprehensively profiling the expression of EBV miRNAs in EBVaGC tissues, EBV-miR-BART4–5p was found to play a role in gastric carcinogenesis through apoptosis regulation by
suppressing the proapoptotic protein Bid (the BH3-interacting domain death agonist) [138]. MiR-BART20–5p contributes to tumorigenesis of EBVaGC by directly interacting with 3′UTR of BAD
[139]. Different from proteins, EBV-microRNAs could escape immune recognition as well as inhibit the immune response by directly suppressing the function of some antiviral host factors to
facilitate the establishment of latent EBV infection. For example, EBV miRNA BART16 have been reported to suppress type I IFN signaling [140]. The oncogenic proteins and miR-BARTs in EBVaGC
were summarized in Table 1. GENETIC AND EPIGENETIC ABNORMALITIES OF HOST CELLS IN EBVAGC Multiple abnormalities of the EBVaGC cells have been identified. Among them, high frequency and
nonrandom DNA methylation attract most attentions [141, 142]. However, the mechanisms are not fully elucidated yet. LMP2A was confirmed to mediate this process. LMP2A induces the STAT3
phosphorylation followed by DNMT1 transcriptionally activation and _PTEN_ promoter methylation, indicating LMP2A plays an essential role in the development and maintenance of EBV-associated
cancer [143]. Besides, a resistance factor against DNA methylation namely TET2 was suppressed to contribute to DNA methylation acquisition during EBV infection [144]. Variety of
tumor-suppressor genes have been identified to be methylated during EBV infection, such as _p16_, _p14_, _APC_, _SSTR1_, _FHIT_, _CRBP1_, _WWOX_, _DLC-1_, _AQP3_, _REC8_, _TP73_, _BLU_,
_FSD1_, _BCL7A_, _MARK1_, _SCRN1_, and _NKX3.1_ [129, 145,146,147,148,149,150,151]. The developed high-throughput sequencing makes it possible to reveal the EBV-induced DNA hypermethylation
comprehensively. Using methyl-DNA immunoprecipitation microarray assays, Zhao et al. found 886 genes involved in cancer-related pathways were aberrantly promoter-hypermethylated in
EBV-positive AGS cells [152]. They also employed whole-genome, transcriptome, and epigenome sequence analyses of EBV-infected or noninfected AGS cells together with primary samples to
comprehensively reveal that EBV infection alters host gene expression through methylation and affects five prominent networks [153]. Apart from the methylation of host cells, EBV could
promote vasculogenic mimicry formation, a new tumor vascular paradigm independent of endothelial cells, in NPC and GC cells through the PI3K/AKT/mTOR/HIF-1α axis [154]. EBV infects ~95% of
people, however only part of the population develops tumors, indicating that molecular abnormalities of host cells are also equally important in the EBV-associated tumorigenesis. As for
EBVaGC, high-frequency mutations of _PIK3CA_, _ARID1A_, and _BCOR_ have been identified. Interestingly, _TP53_ mutation, which counts the most frequent mutation type in cancers, is extremely
rare [113]. The amplification of _JAK2_, _PD-L1_, and _PD-L2_ were also revealed as prominent molecular features [155, 156]. HOST IMMUNITY IN EBV-POSITIVE GC By using gene expression
profile analysis, it was found that the prominent changes in EBVaGCs are immune response genes, which might allow EBVaGC to recruit reactive immune cells [111]. In fact, EBVaGC is
characterized with the high density of CD8+ T cells and low density of CD204+ macrophages [112, 157, 158]. The robust present of infiltrating immune cells and specific microenvironments
partially contribute to antitumor immunity [159]. However, the tumor cells in EBVaGC evade the immune response through multiple strategies. It was reported that indoleamine 2,3-dioxygenase
(IDO1), a potent immune-inhibitory molecule, was upregulated in EBVaGC to resistance tumor immune response [130, 160]. In addition, Tregs were recruited by CCL22 produced by EBVaGC cells to
counteract the antitumor response of CD8+ T cells [161]. EBVaGC also exhibits higher levels of programmed death ligand 1 (PD-L1) expression in carcinoma cells and the infiltrated immune
cells [162, 163]. As tumor cells employ PD-L1 to evade antitumor immunity through interaction with programmed cell death protein 1 on the surface of T cells, the high expression of PD-L1 in
EBVaGC is thought to contribute to the tumor progression [164]. THE DIAGNOSIS AND TREATMENT OF EBVAGC By measuring immune-related proteins in plasma of patients with EBV-positive tumors and
EBV-negative tumors, Camargo et al. found some chemokines and PD-L1 in plasma that could be used for the diagnosis of EBV status [165]. The plasma EBV-DNA load in EBVaGC patients decreases
when the patients show response to the treatment, while load increases when the disease progresses, suggesting that plasma EBV-DNA serves as an ideal marker in predicting recurrence and
chemotherapy response [166]. EBVaGC, MSI-high GC, intestinal type GC as a surrogate for chromosomal instability, diffuse type as a surrogate for genomically stable was classified as four
different subtypes of GC proposed by TCGA [113]. The molecular subtypes of GC are also correlated with the immune subtype [167, 168], suggesting the TCGA classification could be further
employed in future immunotherapy trials. The ACRG classification also revealed four molecular subtypes with clinical outcome. MSI subtype has the best prognosis and lowest recurrence rate
followed by MSS/TP53+ and MSS/TP53−, while the MSS/EMT subtype demonstrates the worst prognosis and highest recurrence rate among the four subtypes. In ACRG classification, EBVaGCs are more
frequently found in the MSS/TP53+ group than in the other groups, indicating a modest survival and recurrence [5]. Patients with EBV-positive tumors showed high responses to pembrolizumab
treatment in a phase II trial of metastatic GC [169]. The satisfied response might rely on that EBVaGC expresses high levels of PD-L1 [165, 170] and exhibits more tumor infiltrating
lymphocytes (TILs) [163, 167, 171, 172]. The amount of TILs has been reported to be associated with improved overall survival in GC patients [173]. In a research of advanced GC patients
treated with nivolumab, only 25% of patients (1/4) demonstrated good response, and this might be because not all EBV-positive tumors show high PD-L1 expression [174]. Evaluating both EBV
status and PD-L1 expression is necessary for predicting clinical benefit of anti-PD-L1 therapy [175]. To some extent, the result indicates that EBV is a potential biomarker for selecting
patients with better response to PD-L1 treatment [176]. In addition to PD-L1, Kim et al. combined PI3K/mTOR dual inhibitor CMG002, together with the autophagy inhibitor CQ, to provide
enhanced therapeutic efficacy against EBVaGC [177]. FUNGUS IN GASTRIC CARCINOGENESIS Fungus is a kind of eukaryotic microorganism, which is widely distributed worldwide. It was identified
that more than 400 species of fungus associated with human beings. These years, the incidence of invasive fungal infections has experienced a dramatic increase globally. Fungus is detectable
in the digestive tract of about 70% of healthy adults in an analysis by using culture dependent methods. Most of them belong to Candida genus, and the number of fungus in the human stomach
is 0–102 CFU/mL [178, 179]. Another research using PCR amplification of bacterial 16S ribosomal RNA genes and fungal internal transcribed spacers identified two fungal genera, _Candida_ and
_Phialemonium_, in gastric fluid from 25 clinically patients [180]. Generally, host immune system could tolerate fungus colonization and defend its invasion. However, the infection will
occur when the balance is disturbed by systemic immunosuppressive such as the acquired immune deficiency syndrome, leukemia and HSCT, solid organ transplantation and immunosuppressant
therapy, anti-microbial and steroid treatments, total parenteral nutrition, iatrogenic catheters and mechanical ventilation, malignant tumors, chemoradiotherapy, and diabetes mellitus [181,
182]. Besides, GI mucosal lesions and surgical procedures can also lead to GI fungal infection [181]. In a gastro-esophageal candidiasis detection by histological examination of biopsies
from 465 patients, it was thought that the candidiasis is usually secondary to mucosal damage [183]. Candidiasis was detected in 54.2% of the gastric ulcer cases and 10.3% of the chronic
gastritis cases. As for GC, the candidiasis was present in 20% of patients [179, 183]. Although the infection of fungal microorganisms in GC is only in rare cases, it is necessary to
eliminate opportunistic infection of Candida to reduce the significant morbidity and mortality. FUTURE DIRECTIONS Although EBV-related and _H. polyri_-related GCs are classified into
different categories, it should be reminded that the stomach is an organ with multiple microorganism coexistence, which means that disease is promoted by multiple microorganisms. In fact,
apart from direct promoting gastric carcinogenesis, _H. pylori_ potentiates the transformation of the gastric mucosa into a hypochlorhidric environment, which further allow other microbes to
colonize. In addition, coinfection with EBV and _H. pylori_ in pediatric patients are associated with more severe inflammation than those with _H. pylori_ infection alone [184]. Although
the underlying mechanism has been partially suggested, such as host SHP1 phosphatase, antagonist of CagA, is downregulated by EBV-induced promoter hypermethylation [185], the synergistic
oncogenic effects of two or more infectious agents remain to be further explored in the future studies. In recent years, the researches about the microbiota in gastrointestinal attract more
and more attentions. However, the studies on the virome and fungus in stomach cancer are still in infancy. As enormous viruses and fungi do exist in human body including our gastrointestinal
tract, it is imperative to understand the relationship between virome/fungi infection and stomach health. REFERENCES * Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh
JW, Comber H, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–403. CAS PubMed Google Scholar * Ferro A, Peleteiro
B, Malvezzi M, Bosetti C, Bertuccio P, Levi F, et al. Worldwide trends in gastric cancer mortality (1980-2011), with predictions to 2015, and incidence by subtype. Eur J Cancer.
2014;50:1330–44. PubMed Google Scholar * Colquhoun A, Arnold M, Ferlay J, Goodman KJ, Forman D, Soerjomataram I. Global patterns of cardia and non-cardia gastric cancer incidence in 2012.
Gut. 2015;64:1881–8. CAS PubMed Google Scholar * Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. *
Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21:449–56.
CAS PubMed Google Scholar * Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388:2654–64. PubMed Google Scholar * de Martel C, Ferlay J,
Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–15. PubMed Google
Scholar * Plummer M, de Martel C, Vignat J, Ferlay J, Bray F, Franceschi S. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob Health.
2016;4:e609–e616. PubMed Google Scholar * Blaser MJ. Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. 2006;7:956–60. CAS PubMed PubMed Central Google Scholar
* Perry S, de la Luz Sanchez M, Yang S, Haggerty TD, Hurst P, Perez-Perez G, et al. Gastroenteritis and transmission of _Helicobacter pylori_ infection in households. Emerg Infect Dis.
2006;12:1701–8. PubMed PubMed Central Google Scholar * Stoicov C, Saffari R, Cai X, Hasyagar C, Houghton J. Molecular biology of gastric cancer: _Helicobacter_ infection and gastric
adenocarcinoma: bacterial and host factors responsible for altered growth signaling. Gene. 2004;341:1–17. CAS PubMed Google Scholar * Uemura N, Okamoto S, Yamamoto S, Matsumura N,
Yamaguchi S, Yamakido M, et al. _Helicobacter pylori_ infection and the development of gastric cancer. N Engl J Med. 2001;345:784–9. CAS PubMed Google Scholar * Wong BC, Lam SK, Wong WM,
Chen JS, Zheng TT, Feng RE, et al. _Helicobacter pylori_ eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291:187–94. CAS
PubMed Google Scholar * Shikata K, Kiyohara Y, Kubo M, Yonemoto K, Ninomiya T, Shirota T, et al. A prospective study of dietary salt intake and gastric cancer incidence in a defined
Japanese population: the Hisayama study. Int J Cancer. 2006;119:196–201. CAS PubMed Google Scholar * Shikata K, Doi Y, Yonemoto K, Arima H, Ninomiya T, Kubo M, et al. Population-based
prospective study of the combined influence of cigarette smoking and _Helicobacter pylori_ infection on gastric cancer incidence: the Hisayama Study. Am J Epidemiol. 2008;168:1409–15. PubMed
Google Scholar * Huang JQ, Zheng GF, Sumanac K, Irvine EJ, Hunt RH. Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology. 2003;125:1636–44.
PubMed Google Scholar * Enroth H, Kraaz W, Engstrand L, Nyren O, Rohan T. _Helicobacter pylori_ strain types and risk of gastric cancer: a case-control study. Cancer Epidemiol, Biomark
Prev. 2000;9:981–5. CAS Google Scholar * Rudi J, Kolb C, Maiwald M, Zuna I, von Herbay A, Galle PR, et al. Serum antibodies against _Helicobacter pylori_ proteins VacA and CagA are
associated with increased risk for gastric adenocarcinoma. Dig Dis Sci. 1997;42:1652–9. CAS PubMed Google Scholar * Yamaoka Y, Kodama T, Kashima K, Graham DY. Antibody against
_Helicobacter pylori_ CagA and VacA and the risk for gastric cancer. J Clin Pathol. 1999;52:215–8. CAS PubMed PubMed Central Google Scholar * Parsonnet J, Friedman GD, Orentreich N,
Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative _Helicobacter pylori_ infection. Gut. 1997;40:297–301. CAS PubMed PubMed Central Google Scholar *
Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic expression of _Helicobacter pylori_ CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl
Acad Sci USA. 2008;105:1003–8. CAS PubMed Google Scholar * Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter exploits integrin for type IV secretion and kinase
activation. Nature. 2007;449:862–6. CAS PubMed Google Scholar * Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, et al. _Helicobacter pylori_ CagA induces Ras-independent
morphogenetic response through SHP-2 recruitment and activation. J Biol Chem. 2004;279:17205–16. CAS PubMed Google Scholar * Tsutsumi R, Takahashi A, Azuma T, Higashi H, Hatakeyama M.
Focal adhesion kinase is a substrate and downstream effector of SHP-2 complexed with _Helicobacter pylori_ CagA. Mol Cell Biol. 2006;26:261–76. CAS PubMed PubMed Central Google Scholar *
Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. _Helicobacter pylori_ CagA interacts with E-cadherin and deregulates the beta-catenin signal that
promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26:4617–26. CAS PubMed Google Scholar * Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, Morikawa
H, et al. _Helicobacter pylori_ CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe. 2009;5:23–34. CAS PubMed Google Scholar *
Jackson CB, Judd LM, Menheniott TR, Kronborg I, Dow C, Yeomans ND, et al. Augmented gp130-mediated cytokine signalling accompanies human gastric cancer progression. J Pathol.
2007;213:140–51. CAS PubMed Google Scholar * Bronte-Tinkew DM, Terebiznik M, Franco A, Ang M, Ahn D, Mimuro H, et al. _Helicobacter pylori_ cytotoxin-associated gene A activates the
signal transducer and activator of transcription 3 pathway in vitro and in vivo. Cancer Res. 2009;69:632–9. CAS PubMed PubMed Central Google Scholar * Menheniott TR, Judd LM, Giraud AS.
STAT3: a critical component in the response to _Helicobacter pylori_ infection. Cell Microbiol. 2015;17:1570–82. CAS PubMed Google Scholar * Knorr J, Ricci V, Hatakeyama M, Backert S.
Classification of _Helicobacter pylori_ virulence factors: is CagA a toxin or not? Trends Microbiol. 2019;27:731–8. * Nagy TA, Frey MR, Yan F, Israel DA, Polk DB, Peek RM Jr. _Helicobacter
pylori_ regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J Infect Dis. 2009;199:641–51. CAS PubMed PubMed Central Google Scholar *
Yamaoka Y. Mechanisms of disease: _Helicobacter pylori_ virulence factors. Nat Rev Gastroenterol Hepatol. 2010;7:629–41. CAS PubMed PubMed Central Google Scholar * Nakayama M, Hisatsune
J, Yamasaki E, Isomoto H, Kurazono H, Hatakeyama M, et al. _Helicobacter pylori_ VacA-induced inhibition of GSK3 through the PI3K/Akt signaling pathway. J Biol Chem. 2009;284:1612–9. CAS
PubMed PubMed Central Google Scholar * Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, et al. The complete genome sequence of the gastric pathogen _Helicobacter
pylori_. Nature. 1997;388:539–47. CAS PubMed Google Scholar * Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, et al. Genomic-sequence comparison of two unrelated isolates of the
human gastric pathogen _Helicobacter pylori_. Nature. 1999;397:176–80. PubMed Google Scholar * Alm RA, Bina J, Andrews BM, Doig P, Hancock RE, Trust TJ. Comparative genomics of
_Helicobacter pylori_: analysis of the outer membrane protein families. Infect Immun. 2000;68:4155–68. CAS PubMed PubMed Central Google Scholar * Yamaoka Y, Kwon DH, Graham DY. A M(r)
34,000 proinflammatory outer membrane protein (oipA) of _Helicobacter pylori_. Proc Natl Acad Sci USA. 2000;97:7533–8. CAS PubMed Google Scholar * Yamaoka Y, Kikuchi S, el-Zimaity HM,
Gutierrez O, Osato MS, Graham DY. Importance of _Helicobacter pylori_ oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology.
2002;123:414–24. CAS PubMed Google Scholar * Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA, et al. Regulation of gastric carcinogenesis by _Helicobacter pylori_
virulence factors. Cancer Res. 2008;68:379–87. CAS PubMed PubMed Central Google Scholar * El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, et al. Interleukin-1
polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. CAS PubMed Google Scholar * El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg
JB, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124:1193–201. CAS PubMed Google Scholar * Hwang
IR, Kodama T, Kikuchi S, Sakai K, Peterson LE, Graham DY, et al. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in _Helicobacter pylori_ infection.
Gastroenterology. 2002;123:1793–803. CAS PubMed Google Scholar * Ando T, Ishikawa T, Kato H, Yoshida N, Naito Y, Kokura S, et al. Synergistic effect of HLA class II loci and cytokine gene
polymorphisms on the risk of gastric cancer in Japanese patients with _Helicobacter pylori_ infection. Int J Cancer. 2009;125:2595–602. CAS PubMed Google Scholar * Lamb A, Chen LF. Role
of the _Helicobacter pylori_-induced inflammatory response in the development of gastric cancer. J Cell Biochem. 2013;114:491–7. CAS PubMed PubMed Central Google Scholar * Beales IL,
Calam J. _Helicobacter pylori_ stimulates granulocyte-macrophage colony-stimulating factor (GM-CSF) production from cultured antral biopsies and a human gastric epithelial cell line. Eur J
Gastroenterol Hepatol. 1997;9:451–5. CAS PubMed Google Scholar * Cho SO, Lim JW, Kim KH, Kim H. Involvement of Ras and AP-1 in _Helicobacter pylori_-induced expression of COX-2 and iNOS
in gastric epithelial AGS cells. Dig Dis Sci. 2010;55:988–96. CAS PubMed Google Scholar * Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, et al. Inflammatory processes
triggered by _Helicobacter pylori_ infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010;70:1430–40. CAS PubMed Google Scholar * Yamashita S, Nanjo S,
Rehnberg E, Iida N, Takeshima H, Ando T, et al. Distinct DNA methylation targets by aging and chronic inflammation: a pilot study using gastric mucosa infected with _Helicobacter pylori_.
Clin Epigenetics. 2019;11:191. CAS PubMed PubMed Central Google Scholar * Leung WK, Man EP, Yu J, Go MY, To KF, Yamaoka Y, et al. Effects of _Helicobacter pylori_ eradication on
methylation status of E-cadherin gene in noncancerous stomach. Clin Cancer Res. 2006;12:3216–21. CAS PubMed Google Scholar * Bimczok D, Clements RH, Waites KB, Novak L, Eckhoff DE, Mannon
PJ, et al. Human primary gastric dendritic cells induce a Th1 response to _H. pylori_. Mucosal Immunol. 2010;3:260–9. CAS PubMed PubMed Central Google Scholar * Ye G, Barrera C, Fan X,
Gourley WK, Crowe SE, Ernst PB, et al. Expression of B7-1 and B7-2 costimulatory molecules by human gastric epithelial cells: potential role in CD4+ T cell activation during _Helicobacter
pylori_ infection. J Clin Investig. 1997;99:1628–36. CAS PubMed Google Scholar * D’Elios MM, Manghetti M, De Carli M, Costa F, Baldari CT, Burroni D, et al. T helper 1 effector cells
specific for _Helicobacter pylori_ in the gastric antrum of patients with peptic ulcer disease. J Immunol. 1997;158:962–7. PubMed Google Scholar * Hafsi N, Voland P, Schwendy S, Rad R,
Reindl W, Gerhard M, et al. Human dendritic cells respond to _Helicobacter pylori_, promoting NK cell and Th1-effector responses in vitro. J Immunol. 2004;173:1249–57. CAS PubMed Google
Scholar * DeLyria ES, Redline RW, Blanchard TG. Vaccination of mice against _H. pylori_ induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterology.
2009;136:247–56. CAS PubMed Google Scholar * Wolfe RR, Jahoor F. Recovery of labeled CO2 during the infusion of C-1- vs C-2-labeled acetate: implications for tracer studies of substrate
oxidation. Am J Clin Nutr. 1990;51:248–52. CAS PubMed Google Scholar * Zhuang Y, Shi Y, Liu XF, Zhang JY, Liu T, Fan X, et al. _Helicobacter pylori_-infected macrophages induce Th17 cell
differentiation. Immunobiology. 2011;216:200–7. CAS PubMed Google Scholar * Eaton KA, Mefford M, Thevenot T. The role of T cell subsets and cytokines in the pathogenesis of _Helicobacter
pylori_ gastritis in mice. J Immunol. 2001;166:7456–61. CAS PubMed Google Scholar * Eaton KA, Mefford ME. Cure of _Helicobacter pylori_ infection and resolution of gastritis by adoptive
transfer of splenocytes in mice. Infect Immun. 2001;69:1025–31. CAS PubMed PubMed Central Google Scholar * Akhiani AA, Pappo J, Kabok Z, Schon K, Gao W, Franzen LE, et al. Protection
against _Helicobacter pylori_ infection following immunization is IL-12-dependent and mediated by Th1 cells. J Immunol. 2002;169:6977–84. CAS PubMed Google Scholar * Stoicov C, Fan X, Liu
JH, Bowen G, Whary M, Kurt-Jones E, et al. T-bet knockout prevents _Helicobacter felis_-induced gastric cancer. J Immunol. 2009;183:642–9. CAS PubMed PubMed Central Google Scholar *
Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS. _Helicobacter pylori_-specific CD4+ CD25 high regulatory T cells suppress memory T-cell responses to _H. pylori_ in infected
individuals. Infect Immun. 2003;71:1755–62. CAS PubMed PubMed Central Google Scholar * Robinson K, Kenefeck R, Pidgeon EL, Shakib S, Patel S, Polson RJ, et al. _Helicobacter
pylori_-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut. 2008;57:1375–85. CAS PubMed Google Scholar * Akhiani AA, Schon K, Franzen LE, Pappo J,
Lycke N. _Helicobacter pylori_-specific antibodies impair the development of gastritis, facilitate bacterial colonization, and counteract resistance against infection. J Immunol.
2004;172:5024–33. CAS PubMed Google Scholar * Kamboj AK, Cotter TG, Oxentenko AS. _Helicobacter pylori_: the past, present, and future in management. Mayo Clin Proc. 2017;92:599–604.
PubMed Google Scholar * Wroblewski LE, Peek RM Jr., Wilson KT. _Helicobacter pylori_ and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23:713–39. CAS PubMed
PubMed Central Google Scholar * Fallone CA, Chiba N, van Zanten SV, Fischbach L, Gisbert JP, Hunt RH, et al. The Toronto Consensus for the Treatment of _Helicobacter pylori_ Infection in
Adults. Gastroenterology. 2016;151:51–69 e14. PubMed Google Scholar * Malfertheiner P, Megraud F, O’Morain CA, Gisbert JP, Kuipers EJ, Axon AT, et al. Management of _Helicobacter pylori_
infection-the Maastricht V/Florence Consensus Report. Gut. 2017;66:6–30. CAS PubMed Google Scholar * Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, et al. Molecular
analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA. 2006;103:732–7. CAS PubMed Google Scholar * Li XX, Wong GL, To KF, Wong VW, Lai LH, Chow DK, et al.
Bacterial microbiota profiling in gastritis without _Helicobacter pylori_ infection or non-steroidal anti-inflammatory drug use. PLoS ONE. 2009;4:e7985. PubMed PubMed Central Google
Scholar * Delgado S, Cabrera-Rubio R, Mira A, Suarez A, Mayo B. Microbiological survey of the human gastric ecosystem using culturing and pyrosequencing methods. Microb Ecol.
2013;65:763–72. CAS PubMed Google Scholar * Coker OO, Dai Z, Nie Y, Zhao G, Cao L, Nakatsu G, et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut. 2018;67:1024–32. CAS
PubMed Google Scholar * Vinasco K, Mitchell HM, Kaakoush NO, Castano-Rodriguez N. Microbial carcinogenesis: Lactic acid bacteria in gastric cancer. Biochimica et Biophysica Acta Rev
Cancer. 2019;1872:188309. CAS Google Scholar * Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, Karaoz U, Contreras M, Blaser MJ, et al. Structure of the human gastric bacterial
community in relation to _Helicobacter pylori_ status. ISME J. 2011;5:574–9. CAS PubMed Google Scholar * Gao JJ, Zhang Y, Gerhard M, Mejias-Luque R, Zhang L, Vieth M, et al. Association
between gut microbiota and _Helicobacter pylori_-related gastric lesions in a high-risk population of gastric cancer. Front Cell Infect Microbiol. 2018;8:202. PubMed PubMed Central Google
Scholar * Tan MP, Kaparakis M, Galic M, Pedersen J, Pearse M, Wijburg OL, et al. Chronic _Helicobacter pylori_ infection does not significantly alter the microbiota of the murine stomach.
Appl Environ Microbiol. 2007;73:1010–3. CAS PubMed Google Scholar * Khosravi Y, Dieye Y, Poh BH, Ng CG, Loke MF, Goh KL, et al. Culturable bacterial microbiota of the stomach of
_Helicobacter pylori_ positive and negative gastric disease patients. Sci World J. 2014;2014:610421. Google Scholar * Nardone G, Compare D. The human gastric microbiota: is it time to
rethink the pathogenesis of stomach diseases. U Eur Gastroenterol J. 2015;3:255–60. CAS Google Scholar * Engstrand L, Lindberg M. _Helicobacter pylori_ and the gastric microbiota. Best
Pract Res Clin Gastroenterol. 2013;27:39–45. CAS PubMed Google Scholar * Virgin HW. The virome in mammalian physiology and disease. Cell. 2014;157:142–50. CAS PubMed PubMed Central
Google Scholar * Cantalupo PG, Katz JP, Pipas JM. Viral sequences in human cancer. Virology. 2018;513:208–16. CAS PubMed Google Scholar * Lieberman PM. Virology. Epstein-Barr virus turns
50. Science. 2014;343:1323–5. CAS PubMed PubMed Central Google Scholar * Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer.
2006;118:3030–44. CAS PubMed Google Scholar * Sokal EM, Hoppenbrouwers K, Vandermeulen C, Moutschen M, Leonard P, Moreels A, et al. Recombinant gp350 vaccine for infectious mononucleosis:
a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis.
2007;196:1749–53. PubMed Google Scholar * Hutt-Fletcher LM. The long and complicated relationship between Epstein-Barr virus and epithelial cells. J Virol. 2017; 91:e01677-16. * Zhang H,
Li Y, Wang HB, Zhang A, Chen ML, Fang ZX, et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat Microbiol. 2018;3:1–8. PubMed Google Scholar *
Chesnokova LS, Hutt-Fletcher LM. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers
a conformational change in glycoproteins gHgL. J Virol. 2011;85:13214–23. CAS PubMed PubMed Central Google Scholar * Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. Fusion of epithelial
cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc Natl Acad Sci USA. 2009;106:20464–9. CAS PubMed
Google Scholar * Xiong D, Du Y, Wang HB, Zhao B, Zhang H, Li Y, et al. Nonmuscle myosin heavy chain IIA mediates Epstein-Barr virus infection of nasopharyngeal epithelial cells. Proc Natl
Acad Sci USA. 2015;112:11036–41. CAS PubMed Google Scholar * Imai S, Nishikawa J, Takada K. Cell-to-cell contact as an efficient mode of Epstein-Barr virus infection of diverse human
epithelial cells. J Virol. 1998;72:4371–8. CAS PubMed PubMed Central Google Scholar * Nanbo A, Kachi K, Yoshiyama H, Ohba Y. Epstein-Barr virus exploits host endocytic machinery for
cell-to-cell viral transmission rather than a virological synapse. J Gen Virol. 2016;97:2989–3006. CAS PubMed Google Scholar * Lieberman PM. Keeping it quiet: chromatin control of
gammaherpesvirus latency. Nat Rev Microbiol. 2013;11:863–75. CAS PubMed PubMed Central Google Scholar * Rowlands DC, Ito M, Mangham DC, Reynolds G, Herbst H, Hallissey MT, et al.
Epstein-Barr virus and carcinomas: rare association of the virus with gastric adenocarcinomas. Br J Cancer. 1993;68:1014–9. CAS PubMed PubMed Central Google Scholar * Imai S, Koizumi S,
Sugiura M, Tokunaga M, Uemura Y, Yamamoto N, et al. Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein-Barr virus latent infection protein. Proc Natl Acad Sci USA.
1994;91:9131–5. CAS PubMed Google Scholar * Ott G, Kirchner T, Muller-Hermelink HK. Monoclonal Epstein-Barr virus genomes but lack of EBV-related protein expression in different types of
gastric carcinoma. Histopathology. 1994;25:323–9. CAS PubMed Google Scholar * Murray PG, Niedobitek G, Kremmer E, Grasser F, Reynolds GM, Cruchley A, et al. In situ detection of the
Epstein-Barr virus-encoded nuclear antigen 1 in oral hairy leukoplakia and virus-associated carcinomas. J Pathol. 1996;178:44–47. CAS PubMed Google Scholar * Sugiura M, Imai S, Tokunaga
M, Koizumi S, Uchizawa M, Okamoto K, et al. Transcriptional analysis of Epstein-Barr virus gene expression in EBV-positive gastric carcinoma: unique viral latency in the tumour cells. Br J
Cancer. 1996;74:625–31. CAS PubMed PubMed Central Google Scholar * Burke AP, Yen TS, Shekitka KM, Sobin LH. Lymphoepithelial carcinoma of the stomach with Epstein-Barr virus demonstrated
by polymerase chain reaction. Mod Pathol. 1990;3:377–80. CAS PubMed Google Scholar * Tokunaga M, Land CE, Uemura Y, Tokudome T, Tanaka S, Sato E. Epstein-Barr virus in gastric carcinoma.
Am J Pathol. 1993;143:1250–4. CAS PubMed PubMed Central Google Scholar * Shibata D, Weiss LM. Epstein-Barr virus-associated gastric adenocarcinoma. Am J Pathol. 1992;140:769–74. CAS
PubMed PubMed Central Google Scholar * Leoncini L, Vindigni C, Megha T, Funto I, Pacenti L, Musaro M, et al. Epstein-Barr virus and gastric cancer: data and unanswered questions. Int J
Cancer. 1993;53:898–901. CAS PubMed Google Scholar * Shibata D, Hawes D, Stemmermann GN, Weiss LM. Epstein-Barr virus-associated gastric adenocarcinoma among Japanese Americans in Hawaii.
Cancer Epidemiol, Biomark Prev. 1993;2:213–7. CAS Google Scholar * Harn HJ, Chang JY, Wang MW, Ho LI, Lee HS, Chiang JH, et al. Epstein-Barr virus-associated gastric adenocarcinoma in
Taiwan. Hum Pathol. 1995;26:267–71. CAS PubMed Google Scholar * Yuen ST, Chung LP, Leung SY, Luk IS, Chan SY, Ho J. In situ detection of Epstein-Barr virus in gastric and colorectal
adenocarcinomas. Am J Surg Pathol. 1994;18:1158–63. CAS PubMed Google Scholar * Gulley ML, Pulitzer DR, Eagan PA, Schneider BG. Epstein-Barr virus infection is an early event in gastric
carcinogenesis and is independent of bcl-2 expression and p53 accumulation. Hum Pathol. 1996;27:20–27. CAS PubMed Google Scholar * Khan G, Hashim MJ. Global burden of deaths from
Epstein-Barr virus attributable malignancies 1990-2010. Infect Agents Cancer. 2014;9:38. PubMed PubMed Central Google Scholar * Murphy G, Pfeiffer R, Camargo MC, Rabkin CS. Meta-analysis
shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology. 2009;137:824–33. PubMed PubMed Central Google Scholar *
Schuster V, Ott G, Seidenspinner S, Kreth HW. Common Epstein-Barr virus (EBV) type-1 variant strains in both malignant and benign EBV-associated disorders. Blood. 1996;87:1579–85. CAS
PubMed Google Scholar * Sidagis J, Ueno K, Tokunaga M, Ohyama M, Eizuru Y. Molecular epidemiology of Epstein-Barr virus (EBV) in EBV-related malignancies. Int J Cancer. 1997;72:72–76. CAS
PubMed Google Scholar * Yanai H, Nishikawa J, Mizugaki Y, Shimizu N, Takada K, Matsusaki K, et al. Endoscopic and pathologic features of Epstein-Barr virus-associated gastric carcinoma.
Gastrointest Endosc. 1997;45:236–42. CAS PubMed Google Scholar * Camargo MC, Kim WH, Chiaravalli AM, Kim KM, Corvalan AH, Matsuo K, et al. Improved survival of gastric cancer with tumour
Epstein-Barr virus positivity: an international pooled analysis. Gut. 2014;63:236–43. PubMed Google Scholar * Kim SY, Park C, Kim HJ, Park J, Hwang J, Kim JI, et al. Deregulation of immune
response genes in patients with Epstein-Barr virus-associated gastric cancer and outcomes. Gastroenterology. 2015;148:137–47 e139. CAS PubMed Google Scholar * Ichimura T, Abe H, Morikawa
T, Yamashita H, Ishikawa S, Ushiku T, et al. Low density of CD204-positive M2-type tumor-associated macrophages in Epstein-Barr virus-associated gastric cancer: a clinicopathologic study
with digital image analysis. Hum Pathol. 2016;56:74–80. PubMed Google Scholar * Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature.
2014;513:202–9. Google Scholar * Luo B, Wang Y, Wang XF, Liang H, Yan LP, Huang BH, et al. Expression of Epstein-Barr virus genes in EBV-associated gastric carcinomas. World J
Gastroenterol. 2005;11:629–33. CAS PubMed PubMed Central Google Scholar * Iwakiri D, Eizuru Y, Tokunaga M, Takada K. Autocrine growth of Epstein-Barr virus-positive gastric carcinoma
cells mediated by an Epstein-Barr virus-encoded small RNA. Cancer Res. 2003;63:7062–7. CAS PubMed Google Scholar * Banerjee AS, Pal AD, Banerjee S. Epstein-Barr virus-encoded small
non-coding RNAs induce cancer cell chemoresistance and migration. Virology. 2013;443:294–305. CAS PubMed Google Scholar * Shinozaki A, Sakatani T, Ushiku T, Hino R, Isogai M, Ishikawa S,
et al. Downregulation of microRNA-200 in EBV-associated gastric carcinoma. Cancer Res. 2010;70:4719–27. CAS PubMed Google Scholar * Cheng TC, Hsieh SS, Hsu WL, Chen YF, Ho HH, Sheu LF.
Expression of Epstein-Barr nuclear antigen 1 in gastric carcinoma cells is associated with enhanced tumorigenicity and reduced cisplatin sensitivity. Int J Oncol. 2010;36:151–60. CAS PubMed
Google Scholar * Sivachandran N, Dawson CW, Young LS, Liu FF, Middeldorp J, Frappier L. Contributions of the Epstein-Barr virus EBNA1 protein to gastric carcinoma. J Virol. 2012;86:60–68.
CAS PubMed PubMed Central Google Scholar * Kim SM, Hur DY, Hong SW, Kim JH. EBV-encoded EBNA1 regulates cell viability by modulating miR34a-NOX2-ROS signaling in gastric cancer cells.
Biochem Biophys Res Commun. 2017;494:550–5. CAS PubMed Google Scholar * Minamitani T, Yasui T, Ma Y, Zhou H, Okuzaki D, Tsai CY, et al. Evasion of affinity-based selection in germinal
centers by Epstein-Barr virus LMP2A. Proc Natl Acad Sci USA. 2015;112:11612–7. CAS PubMed Google Scholar * Hino R, Uozaki H, Inoue Y, Shintani Y, Ushiku T, Sakatani T, et al. Survival
advantage of EBV-associated gastric carcinoma: survivin up-regulation by viral latent membrane protein 2A. Cancer Res. 2008;68:1427–35. CAS PubMed Google Scholar * Liu X, Gao Y, Luo B,
Zhao Y. Construction and antiapoptosis activities of recombinant adenoviral expression vector carrying EBV latent membrane protein 2A. Gastroenterol Res Pract. 2011;2011:182832. PubMed
PubMed Central Google Scholar * Pal AD, Basak NP, Banerjee AS, Banerjee S. Epstein-Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated
by Notch signaling pathway. Carcinogenesis. 2014;35:1592–601. CAS PubMed Google Scholar * Deb Pal A, Banerjee S. Epstein-Barr virus latent membrane protein 2A mediated activation of
sonic Hedgehog pathway induces HLA class Ia downregulation in gastric cancer cells. Virology. 2015;484:22–32. CAS PubMed Google Scholar * Fukuda M, Longnecker R. Latent membrane protein
2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J Virol. 2004;78:1697–705. CAS PubMed PubMed Central Google Scholar
* Fukuda M, Longnecker R. Epstein-Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt Pathway. J Virol. 2007;81:9299–306. CAS
PubMed PubMed Central Google Scholar * Louafi F, Martinez-Nunez RT, Sanchez-Elsner T. MicroRNA-155 targets SMAD2 and modulates the response of macrophages to transforming growth
factor-{beta}. J Biol Chem. 2010;285:41328–36. CAS PubMed PubMed Central Google Scholar * Wang J, Liu W, Zhang X, Zhang Y, Xiao H, Luo B. LMP2A induces DNA methylation and expression
repression of AQP3 in EBV-associated gastric carcinoma. Virology. 2019;534:87–95. CAS PubMed Google Scholar * Strong MJ, Xu G, Coco J, Baribault C, Vinay DS, Lacey MR, et al. Differences
in gastric carcinoma microenvironment stratify according to EBV infection intensity: implications for possible immune adjuvant therapy. PLoS Pathog. 2013;9:e1003341. CAS PubMed PubMed
Central Google Scholar * Marquitz AR, Mathur A, Shair KH, Raab-Traub N. Infection of Epstein-Barr virus in a gastric carcinoma cell line induces anchorage independence and global changes
in gene expression. Proc Natl Acad Sci USA. 2012;109:9593–8. CAS PubMed Google Scholar * Marquitz AR, Mathur A, Chugh PE, Dittmer DP, Raab-Traub N. Expression profile of microRNAs in
Epstein-Barr virus-infected AGS gastric carcinoma cells. J Virol. 2014;88:1389–93. PubMed PubMed Central Google Scholar * Tsai CY, Liu YY, Liu KH, Hsu JT, Chen TC, Chiu CT, et al.
Comprehensive profiling of virus microRNAs of Epstein-Barr virus-associated gastric carcinoma: highlighting the interactions of ebv-Bart9 and host tumor cells. J Gastroenterol Hepatol.
2017;32:82–91. CAS PubMed Google Scholar * Wang J, Zheng X, Qin Z, Wei L, Lu Y, Peng Q, et al. Epstein-Barr virus miR-BART3-3p promotes tumorigenesis by regulating the senescence pathway
in gastric cancer. J Biol Chem. 2019;294:4854–66. CAS PubMed PubMed Central Google Scholar * Zheng X, Wang J, Wei L, Peng Q, Gao Y, Fu Y, et al. Epstein-Barr virus microRNA miR-BART5-3p
inhibits p53 expression. J Virol. 2018;92:e01022-18. * Choy EY, Siu KL, Kok KH, Lung RW, Tsang CM, To KF, et al. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell
survival. J Exp Med. 2008;205:2551–60. CAS PubMed PubMed Central Google Scholar * Marquitz AR, Mathur A, Nam CS, Raab-Traub N. The Epstein-Barr virus BART microRNAs target the
pro-apoptotic protein Bim. Virology. 2011;412:392–400. CAS PubMed PubMed Central Google Scholar * Shinozaki-Ushiku A, Kunita A, Isogai M, Hibiya T, Ushiku T, Takada K, et al. Profiling
of virus-encoded microRNAs in Epstein-Barr virus-associated gastric carcinoma and their roles in gastric carcinogenesis. J Virol. 2015;89:5581–91. CAS PubMed PubMed Central Google Scholar
* Kim H, Choi H, Lee SK. Epstein-Barr virus miR-BART20-5p regulates cell proliferation and apoptosis by targeting BAD. Cancer Lett. 2015;356:733–42. CAS PubMed Google Scholar * Hooykaas
MJG, van Gent M, Soppe JA, Kruse E, Boer IGJ, van Leenen D, et al. EBV microRNA BART16 suppresses type I IFN signaling. J Immunol. 2017;198:4062–73. CAS PubMed Google Scholar * Kang GH,
Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, et al. Epstein-Barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island
methylator phenotype-positive gastric carcinoma. Am J Pathol. 2002;160:787–94. CAS PubMed PubMed Central Google Scholar * Matsusaka K, Kaneda A, Nagae G, Ushiku T, Kikuchi Y, Hino R, et
al. Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 2011;71:7187–97. CAS PubMed Google Scholar * Hino R, Uozaki H,
Murakami N, Ushiku T, Shinozaki A, Ishikawa S, et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric
carcinoma. Cancer Res. 2009;69:2766–74. CAS PubMed Google Scholar * Namba-Fukuyo H, Funata S, Matsusaka K, Fukuyo M, Rahmutulla B, Mano Y, et al. TET2 functions as a resistance factor
against DNA methylation acquisition during Epstein-Barr virus infection. Oncotarget. 2016;7:81512–26. PubMed PubMed Central Google Scholar * Geddert H, zur Hausen A, Gabbert HE, Sarbia M.
EBV-infection in cardiac and non-cardiac gastric adenocarcinomas is associated with promoter methylation of p16, p14 and APC, but not hMLH1. Cell Oncol. 2011;34:209–14. CAS Google Scholar
* Zhao J, Liang Q, Cheung KF, Kang W, Dong Y, Lung RW, et al. Somatostatin receptor 1, a novel EBV-associated CpG hypermethylated gene, contributes to the pathogenesis of EBV-associated
gastric cancer. Br J Cancer. 2013;108:2557–64. CAS PubMed PubMed Central Google Scholar * He D, Zhang YW, Zhang NN, Zhou L, Chen JN, Jiang Y, et al. Aberrant gene promoter methylation of
p16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein-Barr virus-associated gastric carcinomas. Med Oncol. 2015;32:92. PubMed Google Scholar * Yu J, Liang Q, Wang J, Wang K, Gao J, Zhang J, et al.
REC8 functions as a tumor suppressor and is epigenetically downregulated in gastric cancer, especially in EBV-positive subtype. Oncogene. 2017;36:182–93. CAS PubMed Google Scholar *
Okada T, Nakamura M, Nishikawa J, Sakai K, Zhang Y, Saito M, et al. Identification of genes specifically methylated in Epstein-Barr virus-associated gastric carcinomas. Cancer Sci.
2013;104:1309–14. CAS PubMed Google Scholar * Ushiku T, Chong JM, Uozaki H, Hino R, Chang MS, Sudo M, et al. p73 gene promoter methylation in Epstein-Barr virus-associated gastric
carcinoma. Int J Cancer. 2007;120:60–66. CAS PubMed Google Scholar * Farrell PJ. Epstein-Barr virus and cancer. Annu Rev Pathol. 2019;14:29–53. CAS PubMed Google Scholar * Zhao J,
Liang Q, Cheung KF, Kang W, Lung RW, Tong JH, et al. Genome-wide identification of Epstein-Barr virus-driven promoter methylation profiles of human genes in gastric cancer cells. Cancer.
2013;119:304–12. CAS PubMed Google Scholar * Liang Q, Yao X, Tang S, Zhang J, Yau TO, Li X, et al. Integrative identification of Epstein-Barr virus-associated mutations and epigenetic
alterations in gastric cancer. Gastroenterology. 2014;147:1350–62 e1354. CAS PubMed Google Scholar * Xiang T, Lin YX, Ma W, Zhang HJ, Chen KM, He GP, et al. Vasculogenic mimicry formation
in EBV-associated epithelial malignancies. Nat Commun. 2018;9:5009. PubMed PubMed Central Google Scholar * Dong M, Wang HY, Zhao XX, Chen JN, Zhang YW, Huang Y, et al. Expression and
prognostic roles of PIK3CA, JAK2, PD-L1, and PD-L2 in Epstein-Barr virus-associated gastric carcinoma. Hum Pathol. 2016;53:25–34. CAS PubMed Google Scholar * Saito R, Abe H, Kunita A,
Yamashita H, Seto Y, Fukayama M. Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1+ immune cells in Epstein-Barr virus-associated gastric cancer: the prognostic
implications. Mod Pathol. 2017;30:427–39. CAS PubMed Google Scholar * van Beek J, zur Hausen A, Snel SN, Berkhof J, Kranenbarg EK, van de Velde CJ, et al. Morphological evidence of an
activated cytotoxic T-cell infiltrate in EBV-positive gastric carcinoma preventing lymph node metastases. Am J Surg Pathol. 2006;30:59–65. PubMed Google Scholar * Saiki Y, Ohtani H, Naito
Y, Miyazawa M, Nagura H. Immunophenotypic characterization of Epstein-Barr virus-associated gastric carcinoma: massive infiltration by proliferating CD8+ T-lymphocytes. Lab Investig.
1996;75:67–76. CAS PubMed Google Scholar * Song HJ, Srivastava A, Lee J, Kim YS, Kim KM, Ki Kang W, et al. Host inflammatory response predicts survival of patients with Epstein-Barr
virus-associated gastric carcinoma. Gastroenterology. 2010;139:84–92 e82. PubMed Google Scholar * Lu S, Wang LJ, Lombardo K, Kwak Y, Kim WH, Resnick MB. Expression of indoleamine 2,
3-dioxygenase 1 (IDO1) and tryptophanyl-tRNA synthetase (WARS) in gastric cancer molecular subtypes. Appl Immunohistochem Mol Morphol. 2019. https://doi.org/10.1097/PAI.0000000000000761. *
Zhang NN, Chen JN, Xiao L, Tang F, Zhang ZG, Zhang YW, et al. Accumulation mechanisms of CD4(+)CD25(+)FOXP3(+) regulatory T cells in EBV-associated gastric carcinoma. Sci Rep. 2015;5:18057.
CAS PubMed PubMed Central Google Scholar * Derks S, Liao X, Chiaravalli AM, Xu X, Camargo MC, Solcia E, et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers.
Oncotarget. 2016;7:32925–32. PubMed PubMed Central Google Scholar * Saito R, Abe H, Kunita A, Yamashita H, Seto Y, Fukayama M. Overexpression and gene amplification of PD-L1 in cancer
cells and PD-L1(+) immune cells in Epstein-Barr virus-associated gastric cancer: the prognostic implications. Mod Pathol. 2017;30:427–39. CAS PubMed Google Scholar * Cho J, Kang MS, Kim
KM. Epstein-Barr virus-associated gastric carcinoma and specific features of the accompanying immune response. J Gastric Cancer. 2016;16:1–7. PubMed PubMed Central Google Scholar *
Camargo MC, Sivins A, Isajevs S, Folkmanis V, Rudzite D, Gulley ML, et al. Associations of Epstein-Barr virus-positive gastric adenocarcinoma with circulating mediators of inflammation and
immune response. Cancers. 2018;10:E284. * Qiu MZ, He CY, Lu SX, Guan WL, Wang F, Wang XJ, et al. Prospective observation: clinical utility of plasma Epstein-Barr virus DNA load in
EBV-associated gastric carcinoma patients. Int J Cancer. 2019;146:272–80. * Kim TS, da Silva E, Coit DG, Tang LH. Intratumoral immune response to gastric cancer varies by molecular and
histologic subtype. Am J Surg Pathol. 2019;43:851–60. PubMed Google Scholar * Kim JY, Kim WG, Kwon CH, Park DY. Differences in immune contextures among different molecular subtypes of
gastric cancer and their prognostic impact. Gastric Cancer. 2019;22:1164–75. * Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of
clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018;24:1449–58. CAS PubMed Google Scholar * Sasaki S, Nishikawa J, Sakai K, Iizasa H, Yoshiyama H, Yanagihara
M, et al. EBV-associated gastric cancer evades T-cell immunity by PD-1/PD-L1 interactions. Gastric Cancer. 2019;22:486–96. CAS PubMed Google Scholar * Panda A, Mehnert JM, Hirshfield KM,
Riedlinger G, Damare S, Saunders T, et al. Immune activation and benefit from avelumab in EBV-positive gastric cancer. J Natl Cancer Inst. 2018;110:316–20. CAS PubMed Google Scholar *
Xing X, Guo J, Ding G, Li B, Dong B, Feng Q, et al. Analysis of PD1, PDL1, PDL2 expression and T cells infiltration in 1014 gastric cancer patients. Oncoimmunology. 2018;7:e1356144. PubMed
Google Scholar * Yu PC, Long D, Liao CC, Zhang S. Association between density of tumor-infiltrating lymphocytes and prognoses of patients with gastric cancer. Medicine. 2018;97:e11387.
PubMed PubMed Central Google Scholar * Kawazoe A, Shitara K, Kuboki Y, Bando H, Kojima T, Yoshino T, et al. Clinicopathological features of 22C3 PD-L1 expression with mismatch repair,
Epstein-Barr virus status, and cancer genome alterations in metastatic gastric cancer. Gastric Cancer. 2019;22:69–76. CAS PubMed Google Scholar * Mishima S, Kawazoe A, Nakamura Y, Sasaki
A, Kotani D, Kuboki Y, et al. Clinicopathological and molecular features of responders to nivolumab for patients with advanced gastric cancer. J Immunother Cancer. 2019;7:24. PubMed PubMed
Central Google Scholar * Koemans WJ, Chalabi M, van Sandick JW, van Dieren JM, Kodach LL. Beyond the PD-L1 horizon: In search for a good biomarker to predict success of immunotherapy in
gastric and esophageal adenocarcinoma. Cancer Lett. 2019;442:279–86. CAS PubMed Google Scholar * Kim MY, Kruger AJ, Jeong JY, Kim J, Shin PK, Kim SY, et al. Combination therapy with a
PI3K/mTOR dual inhibitor and chloroquine enhances synergistic apoptotic cell death in Epstein-Barr virus-infected gastric cancer cells. Mol Cells. 2019;42:448–59. CAS PubMed PubMed Central
Google Scholar * Schulze J, Sonnenborn U. Yeasts in the gut: from commensals to infectious agents. Dtsch Arzteblatt Int. 2009;106:837–42. Google Scholar * Zwolinska-Wcislo M, Budak A,
Bogdal J, Trojanowska D, Stachura J. Fungal colonization of gastric mucosa and its clinical relevance. Med Sci Monit. 2001;7:982–8. CAS PubMed Google Scholar * von Rosenvinge EC, Song Y,
White JR, Maddox C, Blanchard T, Fricke WF. Immune status, antibiotic medication and pH are associated with changes in the stomach fluid microbiota. ISME J. 2013;7:1354–66. Google Scholar *
Wang ZK, Yang YS, Stefka AT, Sun G, Peng LH. Review article: fungal microbiota and digestive diseases. Aliment Pharmacol Ther. 2014;39:751–66. CAS PubMed Google Scholar * Lamps LW, Lai
KK, Milner DA Jr. Fungal infections of the gastrointestinal tract in the immunocompromised host: an update. Adv Anat Pathol. 2014;21:217–27. PubMed PubMed Central Google Scholar * Scott
BB, Jenkins D. Gastro-oesophageal candidiasis. Gut. 1982;23:137–9. CAS PubMed PubMed Central Google Scholar * Cardenas-Mondragon MG, Carreon-Talavera R, Camorlinga-Ponce M, Gomez-Delgado
A, Torres J, Fuentes-Panana EM. Epstein Barr virus and _Helicobacter pylori_ co-infection are positively associated with severe gastritis in pediatric patients. PLoS ONE. 2013;8:e62850. CAS
PubMed PubMed Central Google Scholar * Saju P, Murata-Kamiya N, Hayashi T, Senda Y, Nagase L, Noda S, et al. Host SHP1 phosphatase antagonizes _Helicobacter pylori_ CagA and can be
downregulated by Epstein-Barr virus. Nat Microbiol. 2016;1:16026. CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We acknowledge the technical support from Core Utilities
of Cancer Genomics and Pathobiology of Department of Anatomical and Cellular Pathology, The Chinese University of Hong Kong. FUNDING The current manuscript is supported by Research Grants
Council of the Hong Kong Special Administrative Region, China [Project nos.: CUHK 14100019, 14118518 (for GRF projects)], and CUHK Direct Grant for Research (2018.002) from The Chinese
University of Hong Kong. AUTHOR INFORMATION Author notes * These authors contributed equally: Yanan Zhao, Jinglin Zhang AUTHORS AND AFFILIATIONS * Department of Anatomical and Cellular
Pathology, State Key Laboratory of Translational Oncology, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong SAR, PR China Yanan Zhao, Jinglin Zhang, Ka Fai To &
Wei Kang * Institute of Digestive Disease, State Key Laboratory of Digestive Disease, The Chinese University of Hong Kong, Hong Kong SAR, PR China Yanan Zhao, Jinglin Zhang, Jun Yu, Ka Fai
To & Wei Kang * Li Ka Shing Institute of Health Science, Sir Y.K. Pao Cancer Center, The Chinese University of Hong Kong, Hong Kong SAR, PR China Yanan Zhao, Jinglin Zhang, Ka Fai To
& Wei Kang * School of Biomedical Sciences, The Chinese University of Hong Kong, Hong Kong SAR, PR China Alfred S. L. Cheng * Department of Medicine and Therapeutics, The Chinese
University of Hong Kong, Hong Kong SAR, PR China Jun Yu Authors * Yanan Zhao View author publications You can also search for this author inPubMed Google Scholar * Jinglin Zhang View author
publications You can also search for this author inPubMed Google Scholar * Alfred S. L. Cheng View author publications You can also search for this author inPubMed Google Scholar * Jun Yu
View author publications You can also search for this author inPubMed Google Scholar * Ka Fai To View author publications You can also search for this author inPubMed Google Scholar * Wei
Kang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS WK and KFT provided direction and instruction in preparing this manuscript. YZ and JZ
reviewed the literatures and drafted this manuscript. ASLC and JY reviewed the manuscript and made significant revisions on the drafts. CORRESPONDING AUTHORS Correspondence to Ka Fai To or
Wei Kang. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with
regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Zhao, Y., Zhang, J., Cheng, A.S.L. _et al._ Gastric cancer: genome damaged by bugs.
_Oncogene_ 39, 3427–3442 (2020). https://doi.org/10.1038/s41388-020-1241-4 Download citation * Received: 23 December 2019 * Revised: 18 February 2020 * Accepted: 20 February 2020 *
Published: 02 March 2020 * Issue Date: 23 April 2020 * DOI: https://doi.org/10.1038/s41388-020-1241-4 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Attention Required! | CloudflareVistorias FISCALIZAÇÃO DA PREFEITURA NO CARNAVAL DE BH REGISTRA NÃO USO DE MÁSCARAS COMO A MAIOR IRREGULARIDADE 3/03/22 ...
Page Unavailable - ABC NewsABC NewsVideoLiveShowsShopLog InStream onOops! Page unavailable.This page either does not exist or is currently unavaila...
Lori TrawinskiAREAS OF EXPERTISE Age diversity in the workforce, mortgage lending, foreclosures, reverse mortgages, consumer debt, stu...
The standard for 24/7/365 virtual urgent care | va west palm beach health care | veterans affairsThe need for health care doesn’t stop just because it’s 5:00 p.m., the weekend, or a holiday. In fact, the busiest time ...
Experimental observation of vortex rings in a bulk magnetABSTRACT Vortex rings are remarkably stable structures that occur in a large variety of systems, such as in turbulent ga...
Latests News
Gastric cancer: genome damaged by bugsABSTRACT Gastric cancer (GC) is one of the leading causes of cancer-related death worldwide. The role of the microorgani...
Vikings season 5: floki gestures explained - secret behind the mysteryThe midseason finale saw Floki (played by Gustaf Skarsgard) make a shockping decision as his utopian vision came crashin...
News corp global boss to appear at media diversity inquiry next fridayGlobal head of News Corp, Robert Thomson is set to appear next week at the media diversity in Australia senate hearing, ...
The effect of short-term feeding experiments with 3′-methyl-4-dimethylaminoazobenzene on rat-liver mitochondrial functionAccess through your institution Buy or subscribe This is a preview of subscription content, access via your institution ...
For the love of lonesome georgeAccess through your institution Buy or subscribe _'WANTED: Lady companion for gentleman castaway on desert island. ...