Bcs1l mutations produce fanconi syndrome with developmental disability
Bcs1l mutations produce fanconi syndrome with developmental disability"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
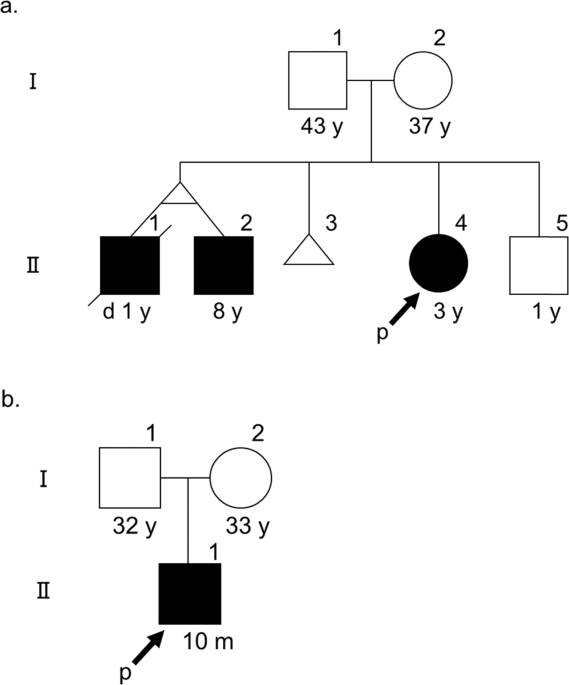
ABSTRACT Fanconi syndrome is a functional disorder of the proximal tubule, characterized by pan-aminoaciduria, glucosuria, hypophosphatemia, and metabolic acidosis. With the advancements in
gene analysis technologies, several causative genes are identified for Fanconi syndrome. Several mitochondrial diseases cause Fanconi syndrome and various systemic symptoms; however, it is
rare that the main clinical symptoms in such disorders are Fanconi syndrome without systematic active diseases like encephalomyopathy or cardiomyopathy. In this study, we analyzed two
families exhibiting Fanconi syndrome, developmental disability and mildly elevated liver enzyme levels. Whole-exome sequencing (WES) detected compound heterozygous known and novel _BCS1L_
mutations, which affect the assembly of mitochondrial respiratory chain complex III, in both cases. The pathogenicity of these mutations has been established in several mitochondria-related
functional analyses in this study. Mitochondrial diseases with isolated renal symptoms are uncommon; however, this study indicates that mitochondrial respiratory chain complex III deficiency
due to _BCS1L_ mutations cause Fanconi syndrome with developmental disability as the primary indications. Access through your institution Buy or subscribe This is a preview of subscription
content, access via your institution ACCESS OPTIONS Access through your institution Subscribe to this journal Receive 12 print issues and online access $259.00 per year only $21.58 per issue
Learn more Buy this article * Purchase on SpringerLink * Instant access to full article PDF Buy now Prices may be subject to local taxes which are calculated during checkout ADDITIONAL
ACCESS OPTIONS: * Log in * Learn about institutional subscriptions * Read our FAQs * Contact customer support SIMILAR CONTENT BEING VIEWED BY OTHERS _COQ7_ DEFECT CAUSES PRENATAL ONSET OF
MITOCHONDRIAL COQ10 DEFICIENCY WITH CARDIOMYOPATHY AND GASTROINTESTINAL OBSTRUCTION Article Open access 03 May 2024 NOVEL _CLTC_ VARIANTS CAUSE NEW BRAIN AND KIDNEY PHENOTYPES Article 07
July 2021 FURTHER DELINEATION OF DEFECTS IN MRPS2 CAUSING HUMAN OXPHOS DEFICIENCY AND EARLY DEVELOPMENTAL ABNORMALITIES IN ZEBRAFISH Article Open access 13 May 2025 REFERENCES * Klootwijk
ED, Reichold M, Unwin RJ, Kleta R, Warth R, Bockenhauer D. Renal Fanconi syndrome: taking a proximal look at the nephron. Nephrol Dial Transpl. 2015;30:1456–60. Article CAS Google Scholar
* Magen D, Berger L, Coady MJ, Ilivitzki A, Militianu D, Tieder M, et al. A loss-of-function mutation in NaPi-IIa and renal Fanconi’s syndrome. N Engl J Med. 2010;362:1102–9. Article CAS
Google Scholar * Klootwijk ED, Reichold M, Helip-Wooley A, Tolaymat A, Broeker C, Robinette SL, et al. Mistargeting of peroxisomal EHHADH and inherited renal Fanconi’s syndrome. N Engl J
Med. 2014;370:129–38. Article CAS Google Scholar * Santer R, Schneppenheim R, Dombrowski A, Götze H, Steinmann B, Schaub J. Mutations in GLUT2, the gene for the liver-type glucose
transporter, in patients with Fanconi-Bickel syndrome. Nat Genet. 1997;17:324–6. Article CAS Google Scholar * Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, et al.
The HNF4A R76W mutation causes atypical dominant Fanconi syndrome in addition to a β cell phenotype. J Med Genet. 2014;51:165–9. Article CAS Google Scholar * Reichold M, Klootwijk ED,
Reinders J, Otto EA, Milani M, Broeker C, et al. Glycine amidinotransferase (GATM), renal Fanconi syndrome, and kidney failure. J Am Soc Nephrol. 2018;29:1849–58. Article CAS Google
Scholar * Niaudet P, Rötig A. Renal involvement in mitochondrial cytopathies. Pediatr Nephrol. 1996;10:368–73. Article CAS Google Scholar * Govers LP, Toka HR, Hariri A, Walsh SB,
Bockenhauer D. Mitochondrial DNA mutations in renal disease: an overview. Pediatr Nephrol. 2021;36:9–17. Article Google Scholar * Murayama K, Shimura M, Liu Z, Okazaki Y, Ohtake A. Recent
topics: the diagnosis, molecular genesis, and treatment of mitochondrial diseases. J Hum Genet. 2019;64:113–25. Article Google Scholar * Kuwertz-Bröking E, Koch HG, Marquardt T, Rossi R,
Helmchen U, Müller-Höcker J, et al. Renal Fanconi syndrome: first sign of partial respiratory chain complex IV deficiency. Pediatr Nephrol. 2000;14:495–8. Article Google Scholar * Morris
AA, Taylor RW, Birch-Machin MA, Jackson MJ, Coulthard MG, Bindoff LA, et al. Neonatal Fanconi syndrome due to deficiency of complex III of the respiratory chain. Pediatr Nephrol.
1995;9:407–11. Article CAS Google Scholar * Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a
joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. Article Google Scholar *
Yatsuga S, Fujita Y, Ishii A, Fukumoto Y, Arahata H, Kakuma T, et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann Neurol. 2015;78:814–23. Article
CAS Google Scholar * Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X, Thorburn DR. Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology.
1999;52:1255–64. Article CAS Google Scholar * Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann
Neurol. 1996;39:343–51. Article CAS Google Scholar * Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and
children. Neurology. 2002;59:1406–11. Article CAS Google Scholar * Van Coster R, Smet J, George E, De Meirleir L, Seneca S, Van Hove J, et al. Blue native polyacrylamide gel
electrophoresis: a powerful tool in diagnosis of oxidative phosphorylation defects. Pediatr Res. 2001;50:658–65. Article Google Scholar * Dabbeni-Sala F, Di Santo S, Franceschini D, Skaper
SD, Giusti P. Melatonin protects against 6-OHDA-induced neurotoxicity in rats: a role for mitochondrial complex I activity. FASEB J. 2001;15:164–70. Article CAS Google Scholar * Calvo
SE, Compton AG, Hershman SG, Lim SC, Lieber DS, Tucker EJ, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med.
2012;4:118ra10. Article Google Scholar * Shigematsu Y, Hayashi R, Yoshida K, Shimizu A, Kubota M, Komori M, et al. Novel heterozygous deletion mutation c.821delC in the AAA domain of BCS1L
underlies Björnstad syndrome. J Dermatol. 2017;44:e111–e12. Article CAS Google Scholar * Cruciat CM, Hell K, Fölsch H, Neupert W, Stuart RA. Bcs1p, an AAA-family member, is a chaperone
for the assembly of the cytochrome bc(1) complex. EMBO J. 1999;18:5226–33. Article CAS Google Scholar * Jackson CB, Bauer MF, Schaller A, Kotzaeridou U, Ferrarini A, Hahn D, et al. A
novel mutation in BCS1L associated with deafness, tubulopathy, growth retardation and microcephaly. Eur J Pediatr. 2016;175:517–25. Article CAS Google Scholar * Ezgu F, Senaca S, Gunduz
M, Tumer L, Hasanoglu A, Tiras U, et al. Severe renal tubulopathy in a newborn due to BCS1L gene mutation: effects of different treatment modalities on the clinical course. Gene.
2013;528:364–6. Article CAS Google Scholar * Baker RA, Priestley JRC, Wilstermann AM, Reese KJ, Mark PR. Clinical spectrum of BCS1L Mitopathies and their underlying structural
relationships. Am J Med Genet A. 2019;179:373–80. Article CAS Google Scholar * Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, et al. COQ2
nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18:2773–80. Article CAS Google Scholar * Heeringa SF, Chernin G, Chaki M,
Zhou W, Sloan AJ, Ji Z, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest. 2011;121:2013–24. Article CAS Google Scholar *
Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest.
2013;123:5179–89. Article CAS Google Scholar * Koga Y, Povalko N, Inoue E, Ishii A, Fujii K, Fujii T, et al. A new diagnostic indication device of a biomarker growth differentiation
factor 15 for mitochondrial diseases: from laboratory to automated inspection. J Inherit Metab Dis. 2021;44:358–66. Article CAS Google Scholar * Invernizzi F, D’Amato I, Jensen PB,
Ravaglia S, Zeviani M, Tiranti V. Microscale oxygraphy reveals OXPHOS impairment in MRC mutant cells. Mitochondrion. 2012;12:328–35. Article CAS Google Scholar * Ogawa E, Shimura M,
Fushimi T, Tajika M, Ichimoto K, Matsunaga A, et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: a study of 106 Japanese patients. J Inherit Metab
Dis. 2017;40:685–93. Article CAS Google Scholar * Arakawa C, Endo A, Kohira R, Fujita Y, Fuchigami T, Mugishima H, et al. Liver-specific mitochondrial respiratory chain complex I
deficiency in fatal influenza encephalopathy. Brain Dev. 2012;34:115–7. Article Google Scholar Download references FUNDING This study was supported by Grants-in-Aid for Scientific Research
(KAKENHI) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (subject ID: 19K17297 to NS, 17H04189 to KI, 19K08726 to KN, and 18K07892 to YK). This study was
also supported partly by the Japan Agency for Medical Research and Development, Grant/Award Numbers: JP17ek0109088 and JP19ek0109336 to YK. AUTHOR INFORMATION Author notes * These authors
contributed equally: Kojima-Ishii Kanako, Nana Sakakibara. AUTHORS AND AFFILIATIONS * Department of Pediatrics, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan
Kojima-Ishii Kanako, Yuko Ichimiya & Yuichi Mushimoto * Department of Pediatrics, Kobe University Graduate School of Medicine, Kobe, Japan Nana Sakakibara, Tomoko Horinouchi, China
Nagano, Tomohiko Yamamura, Kazumoto Iijima & Kandai Nozu * Center for Medical Genetics and Department of Metabolism, Chiba Children’s Hospital, Chiba, Japan Kei Murayama * Department of
Pediatrics, Uwajima City Hospital, Uwajima, Japan Koji Nagatani & Satoshi Murata * Center for Intractable Diseases, Saitama Medical University Hospital, Saitama, Japan Akira Otake *
Department of Pediatrics & Clinical Genomics, Faculty of Medicine, Saitama Medical University, Saitama, Japan Akira Otake * Department of Pediatrics and Child Health, Kurume University
Graduate School of Medicine, Kurume, Japan Yasutoshi Koga * Center for Medical Genetics, Keio University School of Medicine, Tokyo, Japan Hisato Suzuki, Tomoko Uehara & Kenjiro Kosaki *
Department of Human Genetics, Nagasaki University Graduate School of Biomedical Sciences, Atomic Bomb Disease Institute, Nagasaki, Japan Koh-ichiro Yoshiura & Hiroyuki Mishima Authors *
Kojima-Ishii Kanako View author publications You can also search for this author inPubMed Google Scholar * Nana Sakakibara View author publications You can also search for this author
inPubMed Google Scholar * Kei Murayama View author publications You can also search for this author inPubMed Google Scholar * Koji Nagatani View author publications You can also search for
this author inPubMed Google Scholar * Satoshi Murata View author publications You can also search for this author inPubMed Google Scholar * Akira Otake View author publications You can also
search for this author inPubMed Google Scholar * Yasutoshi Koga View author publications You can also search for this author inPubMed Google Scholar * Hisato Suzuki View author publications
You can also search for this author inPubMed Google Scholar * Tomoko Uehara View author publications You can also search for this author inPubMed Google Scholar * Kenjiro Kosaki View author
publications You can also search for this author inPubMed Google Scholar * Koh-ichiro Yoshiura View author publications You can also search for this author inPubMed Google Scholar * Hiroyuki
Mishima View author publications You can also search for this author inPubMed Google Scholar * Yuko Ichimiya View author publications You can also search for this author inPubMed Google
Scholar * Yuichi Mushimoto View author publications You can also search for this author inPubMed Google Scholar * Tomoko Horinouchi View author publications You can also search for this
author inPubMed Google Scholar * China Nagano View author publications You can also search for this author inPubMed Google Scholar * Tomohiko Yamamura View author publications You can also
search for this author inPubMed Google Scholar * Kazumoto Iijima View author publications You can also search for this author inPubMed Google Scholar * Kandai Nozu View author publications
You can also search for this author inPubMed Google Scholar CONTRIBUTIONS K-I K, NS, and KN: conception or design; K-I K, NS, KM, AO, and YK: data collection and analysis; K-I K and NS: data
interpretation. HS, TU, KK, K-I Y, and HM: WES analysis and interpretation; K-I K, KN, SM, YI and YM: collection of patient samples and clinical information; K-I K and NS: drafting the
article; TH, CN, TY, and KI: critical revision of the article. All authors approved the final version of the manuscript for publication. CORRESPONDING AUTHOR Correspondence to Nana
Sakakibara. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICAL APPROVAL All procedures involving human participants were in accordance with the
ethical standards of the Institutional Review Board of Kobe University Graduate School of Medicine (IRB approval number 301), the Kurume University Institutional Review Board (IRB approval
number 273), the Kyushu University Institutional Review Board (IRB approval number 667-00), and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Written informed consent was obtained from all individuals participating in this study. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional
claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY TABLE 1 SUPPLEMENTARY TABLE 2 SUPPLEMENTARY TABLE 3 RIGHTS AND PERMISSIONS Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kanako, KI., Sakakibara, N., Murayama, K. _et al._ _BCS1L_ mutations produce Fanconi syndrome with developmental disability. _J Hum Genet_
67, 143–148 (2022). https://doi.org/10.1038/s10038-021-00984-0 Download citation * Received: 04 April 2021 * Revised: 07 September 2021 * Accepted: 01 October 2021 * Published: 15 October
2021 * Issue Date: March 2022 * DOI: https://doi.org/10.1038/s10038-021-00984-0 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable
link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Garena free fire max redeem codes for april 30, 2022: redeem latest ff reward using codesGarena Free Fire Max is the enhanced version of Garena Free Fire, the adventure-driven battle royale game. Free Fire Max...
186 kilograms of cocaine seized in TasmaniaAdNewsLocal NewsNewsLocal NewsNews HomeNewsSportCommunityTributes & FuneralsClassifiedsExplore TravelEntertainmentLifest...
Thank You, Tom Fuentes - Newport Beach NewsMy father passed away two weeks before my 15th birthday so I’ve spent the past 25 years looking for father figures, ment...
French language tests harden: what changes and how to know your levelRules set to be enforced by a new immigration law in France include stricter language requirements for those applying fo...
Germany Pulls Out a Late VictoryDORTMUND, Germany — There are nights, even at the World Cup, where the stars remain hidden and it is left to the support...
Latests News
Bcs1l mutations produce fanconi syndrome with developmental disabilityABSTRACT Fanconi syndrome is a functional disorder of the proximal tubule, characterized by pan-aminoaciduria, glucosuri...
Haryana cm reviews development projects in gurugram - the statesmanHaryana Chief Minister Nayab Singh Saini, while presiding over a review meeting on the various development projects in G...
Gallery: furniture for the future displayed at design tasmaniaFloyd Drew, 32, created a collection based on his personal experiences. He has drawn on both the physical shapes of the ...
SDYC Wins Youth Regatta, Gov Cup BerthThe undefeated team from SDYC will go to its third Governor’s Cup Challenge in July following a first-place finish in th...
Iris is part of Jamieson familyAdNewsLocal NewsNewsLocal NewsNews HomeNewsSportCommunityTributes & FuneralsClassifiedsExplore TravelEntertainmentLifest...