Identification of the minimal combination of clinical features in probands for efficient mutation detection in the fbn1 gene
Identification of the minimal combination of clinical features in probands for efficient mutation detection in the fbn1 gene"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Mutations identified in the fibrillin-1 (_FBN1_) gene have been associated with Marfan syndrome (MFS). Molecular analysis of the gene is classically performed in probands with MFS
to offer diagnosis for at-risk relatives and in children highly suspected of MFS. However, _FBN1_ gene mutations are found in an ill-defined group of diseases termed ‘type I
fibrillinopathies’, which are associated with an increased risk of aortic dilatation and dissection. Thus, there is growing awareness of the need to identify these non-MFS probands, for
which _FBN1_ gene screening should be performed. To answer this need we compiled the molecular data obtained from the screening of the _FBN1_ gene in 586 probands, which had been addressed
to our laboratory for molecular diagnosis. In this group, the efficacy of _FBN1_ gene screening was high in classical MFS probands (72.5%,), low (58%) in those referred for incomplete MFS
and only slight (14.3%) for patients referred as possible MFS. Using recursive partitioning, we found that the best predictor of the identification of a mutation in the _FBN1_ gene was the
presence of features in at least three organ systems, combining one major, and various minor criteria. We also show that our original recommendation of two systems involved with at least one
with major criterion represents the minimal criteria because in probands not meeting these criteria, the yield of mutation identification drastically falls. This recommendation should help
clinicians and biologists in identifying probands with a high probability of carrying a _FBN1_ gene mutation, and thus optimize biological resources. SIMILAR CONTENT BEING VIEWED BY OTHERS
DEVELOPING A MORE ACCURATE POPULATION FREQUENCY OF MARFAN SYNDROME FROM PREDICTED PATHOGENIC _FBN1_ VARIANTS IN THE GNOMAD COHORTS Article Open access 18 March 2025 GENETIC INSIGHTS FROM A
BRAZILIAN COHORT OF AORTOPATHIES THROUGH TARGETED NEXT-GENERATION SEQUENCING AND _FBN1_ DIRECT SEQUENCING Article Open access 08 November 2024 CLINICALLY RELEVANT VARIANTS IN A LARGE COHORT
OF INDIAN PATIENTS WITH MARFAN SYNDROME AND RELATED DISORDERS IDENTIFIED BY NEXT-GENERATION SEQUENCING Article Open access 12 January 2021 INTRODUCTION Marfan syndrome (MFS, OMIM#154700) is
an inherited autosomal dominant disorder of connective tissue with an estimated incidence of 1/5000 live births with more than 25% sporadic cases. MFS is characterized by a broad range of
clinical manifestations involving the skeletal, ocular, cardiovascular, integument, pulmonary, and central nervous systems with great phenotypic variability.1, 2 Cardiovascular involvement
in the form of aortic aneurysm or dissecting aorta is the most serious life-threatening aspect of the syndrome and can be prevented by timely cardiovascular surgery. Mutations in the gene
encoding fibrillin-1 (_FBN1_, OMIM#134797) cause MFS, as well as other related disorders of connective tissue grouped under the generic term of type-1 fibrillinopathies.3 We have shown that
_FBN1_ mutations are associated with an increased risk of aortic dilatation and dissection, whatever may be the clinical presentation.4 The coding sequence of the _FBN1_ gene is spread over
65 coding exons, and contains 43 calcium binding (cb) epidermal growth factor-like (EGF) modules.5, 6 Private mutations have been identified over the entire length of the gene with no
phenotypic association or apparent clustering in any specific region, with the exception of the severe neonatal form of the syndrome associated with mutations located between exons 24 and
32.7, 8, 9, 10, 11 The detection of a mutation, thus long, difficult and costly, cannot be offered to all suspected Marfan patients even in countries, in which state funding is available for
wide molecular diagnosis. Therefore, we recommended earlier11 that _FBN1_ molecular analysis be performed in newly suspected MFS when two systems are involved with at least one major
diagnostic criterion (as described in the Ghent nosology). In patients found to harbor a mutation, this recommendation allows for diagnosis of MFS. However, _FBN1_ molecular analysis must
also be performed in the ill-defined group of type I fibrillinopathies, as they carry an increased risk of cardiovascular disease.11 Therefore, the question arises of when to perform _FBN1_
gene mutation screening in patients with incomplete phenotypes. To answer this question, we first compiled the molecular data obtained from the screening of the _FBN1_ gene in 586 probands,
which had been addressed to our laboratory for molecular diagnosis. We then performed recursive partitioning on the molecular data obtained for probands, in which the requirements of the
Ghent nosology for diagnosis of MFS were not met. We built a regression tree to identify combinations of clinical features related to mutation rate detection. We found that in patients with
full screening of the Ghent criteria, but dural ectasia (as this feature is not systematically looked for in clinical practice)11 the best predictor of the identification of a mutation in
the _FBN1_ gene was the presence of an ectopia lentis (EL) or features in at least three organ systems, combining one major and various minor criteria. We also found that our original
recommendation of two systems involved at least one with major criterion, represents the minimal criteria because in probands not meeting these criteria, the yield of mutation identification
drastically falls. MATERIALS AND METHODS PATIENTS Blood samples were obtained for probands originating from all over the national territory and referred for molecular diagnosis to our
laboratory (the first French national reference laboratory for MFS) between 1994 and 2006. Informed consent was provided for all patients in agreement with the French bioethic laws. Since
1996, the majority of patients were referred by the Multidisciplinary Marfan Clinic of our University Hospital. There, patients are evaluated by geneticists, rheumatologists or pediatricians
(depending on their age), cardiologists, and ophthalmologists. Systematic slit-lamp examination, cardiac ultrasonography, and radiological investigations are also performed. Dural ectasia
is not systematically searched by magnetic resonance (MR) or computed tomography (CT). For samples referred from other centers, the clinical data of patients were routinely collected before
mutation screening, but complete data were not always available. Patients with missing data (except neurological screening) were not classified and were not included in the statistical
analyses. All samples referred were not screened. Screening exclusion criteria evolved through the years due to a revision of the international diagnostic criteria for MFS,12, 13 but
generally probands with an isolated system involvement were not screened (except for familial cases of EL or aortic aneurysm/dissection). For the purpose of this study, all clinical data
were reassessed by one physician (C.S.), using the Ghent criteria12 to ensure homogeneity of clinical data across centers. In current practice, only readily available clinical data are used
for initial evaluation, and imaging in search of dural ectasia is rarely performed.11 In keeping with this, only 186 of the 586 probands had an investigation of the lumbar spine by CT or MR
imaging. Therefore, we relied mainly on the involvement of the skeletal, ocular, and cardiovascular systems to classify them. Furthermore, the probands tested were subdivided into neonatal
MFS (defined by severe valvular involvement and clinical features of MFS before 4 weeks of age), probable MFS (defined by incomplete Ghent criteria in childhood, that is, <18 years),
incomplete Marfan (patients with incomplete Ghent criteria in adulthood, but with at least one system involved with a major criterion and another system with a minor criterion), thoracic
aortic aneurysm or dissection (TAAD defined as major aortic involvement without any other system involved), EL, or skeletal involvement only, non-MFS (defined by only minor involvement in
one or more systems), and classical MFS (defined by positive Ghent criteria, whatever may be the age). Finally, 18 probands presenting with phenotypes overlapping MFS were also investigated:
Weill-Marchesani syndrome (WMS), Lujan Fryns syndrome, Shprintzen Goldberg syndrome (SGS), Loeys-Dietz syndrome (LDS), Furlong syndrome [(FS) now integrated in the clinical spectrum of LDS
type 1A (OMIM#609192)], or Ehlers-Danlos syndrome (EDS) vascular type. All these disorders were diagnosed using recognized criteria.13, 14, 15, 16, 17, 18 DNA AMPLIFICATION AND MUTATION
DETECTION Genomic DNA was isolated from peripheral blood leukocytes by standard procedures. Initially, all the 65 coding _FBN1_ exons and their splice junctions were amplified using the
primers and conditions described by Nijbroek _et al_8 with the exception of eight newly designed primers for exon 1 (Ex1F: 5′-GAC GGG CGG CGG GAT AGC- 3′; Ex1B: 5′- TGG ATC TTG AAA CTT
GGG-3′), exon 5 (Ex5F: 5′-TTT ATT GTT GTC CTT CCA GAG G-3′; Ex5B: 5′-GCC ATG CAG ACC CAA TGT C-3′), exon 47 (Ex47F: 5′-TAT TAA AGG AAT TGT TGG GG-3′; Ex47B: 5′-TTC CAG GTC TTT CTA AGT
CC-3′), and exon 49 (Ex49F: 5′-TGA TGA TGT CTC CAT CGT GT-3′; Ex49B: 5′-TGC AGC ATT GAA AGC CCA AA-3′). Subsequently, primers specifically designed to amplify all regions at the same
annealing temperature were used (Dr P Khau Van Kien private communication). All PCRs were carried out in the GenAmp PCR System 9700 (Applied Biosystem, Warrington, Cheshire, UK) with reagent
master mix (AB gene). Bidirectional sequencing was performed. Altered exons were resequenced on new DNA dilutions prepared from stock DNA (Big Dye terminators kit, ABI 3100 Genetic
Analyzer, Applied Biosystems). When the mutation altered the regional restriction map, the presence of the mutation was also checked by PCR/digestion using the appropriate restriction
enzyme. PREDICTION OF THE FUNCTIONAL EFFECT OF AMINO ACID SUBSTITUTION To assess the deleterious effect of identified sequence variants, we used various algorithms (Polymorphism Phenotyping
(PolyPhen); Sorting Intolerant From Tolerant (SIFT), Biochemical values and BLOSUM 62), which were developed to predict whether or not a nucleotide variation is likely to affect protein
function. All, except PolyPhen, are implemented in the new version of the UMD-LSDB software in the ‘UMD-Predictor’ tool, kindly provided by Dr C Béroud (manuscript submitted). STATISTICAL
ANALYSIS Qualitative data were compared by the Pearson _χ_2 test, or Fisher’s exact test for small samples. A _P_-value of <0.05 was considered significant. A regression tree was built
through recursive partitioning to identify combinations of clinical features related to mutation detection rate. The algorithm chooses the split on a variable that partitions the data set
into two parts such that it minimizes the sum of the squared deviations from the mean in the separate parts. This splitting or partitioning is then applied to each of the new branches. The
process continues until each node reaches a user-specified minimum node size and becomes a terminal node. Stopping rule was a minimum node size of ten. Trees were cross-validated to prevent
overfitting. Statistical analyses were performed using SPSS software version 12.0 (Chicago, Ill) and R language (R is a language and environment for statistical computing. It was developed
by R Development Core Team (2005); R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL: http://www.R-project.org.). RESULTS MUTATION ANALYSIS In our set of 586
patients screened by direct sequencing, 354 mutations in 345 probands were identified (success rate of 58.9%). It should be noted that nine patients presented on the same allele, two
mutational events predicted as pathogenic. For all probands, the 65 coding exons of the _FBN1_ gene were studied. Molecular data of each identified mutation are summarized in Supplementary
Tables 1–6. Over 200 unrelated French non-MFS patients referred to the laboratory for diagnosis were screened to exclude polymorphisms. In all cases for which family DNA samples were
available, complete cosegregation of the mutational event with the clinically affected status was observed. Among the 354 mutations detailed in this report, two mutations detected in this
cohort have been described elsewhere (c.2954G>A and c.3965-2A>G (c.IVS31-2A>G));19, 20 and 87 mutations have been made available to the community through the UMD-_FBN1_ website
since 2003.21, 22 Mutations are distributed as follows: 196 missense mutations, 48 alterations affecting conserved splice sites, 55 nonsense mutations, 14 insertions/duplications, and 41
deletions. Among the mutations found, 188 (53.1%) were familial. RECURRENT MUTATIONS All mutations identified in the French probands are newly described mutations except 97 different
mutations, which were earlier found in non-French probands (Supplementary Table 1). Among these 97 mutations, 18 were found in more than one French proband. Haplotype analysis, when
possible, showed that the mutations were carried on different chromosomal backgrounds or genotyping of the probands’ parents showed a _de novo_ molecular event, thus excluding the existence
of a French founder mutation. The most frequent mutation reported in the FBN1 database was one of the first recurrent mutations published, c.5788+5G>A (c.IVS46+5G>A)8, 23 reported 18
times. This mutation was found only in two French probands. Conversely, mutation c.7754T>C initially described by Liu _et al_24 was found in five unrelated French families, but only
reported 10 times in the FBN1 database. DOUBLE MUTANTS Nine double mutants have been characterized (c.3299G>T;8176C>T), (c.4143G>C;c.3838+1G>C), (c.442C>T;c.1844delA),
(c.308dup;c.1957_1958dup), (6388G>A;8176C>T), (c.1416C>A;c.8176C>T), (c.986T>C;c.6832C>T), (c.4057G>A;c.2215T>C), and (c.3909delT;c.3188C>G). For all these
probands, parent samples were studied and showed that both mutations were carried on the same allele. Only one case with two mutations has been reported earlier: but for a compound
heterozygote (c.3212T>G;c.3219A>T).25 DELETIONS Forty-one mutations are deletions (suspected deletion mechanism reported in Supplementary Table 2). Thirty-nine result in a premature
termination codon. Two mutations are in-frame: one deletes a cysteine implicated in a disulfide bond, predicting an incorrect folding of the monomer (c.4349_4351delGCT). The second one
deletes two amino acids Pro1352 and Gly1353. The Gly1353 is a highly conserved amino acid in cbEGF-like modules. The c.2420delA mutation concerns the first nucleotide of exon 20. However,
the consensus splice site is very likely not inactivated by the deletion, as the consensus acceptor splice site value is not modified. The mutational mechanism is then likely to be a
frameshift with the appearance of a premature termination codon. INSERTIONS AND DUPLICATIONS Fourteen mutations were insertions (suspected insertion mechanism reported in Supplementary Table
3). Twelve result in a premature termination codon. Two mutations are in-frame and add a cysteine, predicting an incorrect folding of the monomer. SPLICE SITE MUTATIONS Forty-eight
mutations were splice mutations that are predicted to cause abnormal splicing patterns by the use of the nearest and strongest consensus splice site. Thirty-three mutations are located in
the intron at the canonical splice sequences. Thirteen of them inactivate an acceptor splice site and twenty a donor splice site. Thirteen mutations are located in exonic sequences of the
canonical splice sequences in the last nucleotide of the affected exon (Supplementary Table 4). Two mutations create cryptic acceptor splice sites c.5423-28delCCT (c.IVS43-28delCCT) and
c.6997+17delC (c.IVS56+17delC). It is interesting to note that three mutations were located in exonic consensus splice site sequences, but are not likely to modify the used splice site:
c.442C>T implicating the last nucleotide of exon 4 (consensus donor splice site value not modified), c.3209A>G mutation implicating the first nucleotide of exon 26 (consensus acceptor
splice site value increased), and c.6740A>G mutation implicating the first nucleotide of exon 55 (consensus acceptor splice site value increased). The mutational mechanism in these cases
is then most likely an amino acid substitution and these mutations are described in Supplementary Table 6. NONSENSE AND MISSENSE MUTATIONS Fifty-five nonsense mutations have been identified
(Supplementary Table 5). Eight are ocher mutations (TAA), 13 are amber mutations (TAG), and 34 are opal mutations (TGA). One hundred and ninety-six missense mutations have been characterized
(Supplementary Table 6). Neutral variants are differentiated from pathogenic nucleotide substitutions by the use of a new tool implemented in the UMD-database (manuscript submitted). This
tool takes into account the location of the variant at the protein level, that is, in which domain and whether it is involved in a highly conserved domain. The new algorithm implemented in
the UMD-database scored all the mutations we report as pathogenic. CLASSIFICATION ACCORDING TO PREDICTED MUTATION CONSEQUENCES One hundred and fifty-four mutations (representing 42.8% of
mutations) are predicted to result in shortened fibrillin-1 molecules. These mutations are distributed as follows: 55 nonsense, 48 splicing errors, 12 frameshift insertions/duplications, and
39 frameshift deletions. Mutations predicting an incorrect folding of the monomer concern 119 mutations: (1) substitution of a cysteine implicated in a disulfide bond is reported in 93
cases; (2) on the other hand, 22 mutations create a new cysteine potentially disturbing the correct cysteine pairing; and (3) four substitutions of a glycine implicated in correct
domain–domain packing are reported.26 In the other mutations, 42 mutations concern amino acids known to be implicated in Ca2+ binding.27 Fourteen mutations are located at amino acids highly
conserved among species. CLINICAL EVALUATION IN OUR POPULATION The clinical spectrum before _FBN1_ testing and according to data referred by physicians was the following: 21 cases of
neonatal MFS, 21 probable MFS (children), 105 incomplete MFS (adult) (20% of adults), 266 cases of classical MFS (50.5% of adults), 21 non-MFS (4%), and six TAAD, eight EL, one isolated
skeletal involvement only. For 119 patients, investigation of one or more systems (other than dural ectasia) was not available and therefore we did not include them in one of the earlier
clinical groups and in the statistical analysis. Finally, three Lujan Fryns, six SGS, three WMS, one Furlong syndrome, two EDS vascular types (with a _COL3A1_-negative gene screening), and
three LDS were also studied. These 18 probands were not included in the statistical analysis. EFFICACY OF MUTATION SCREENING The overall mutation detection rate was 193/266 (72.5%) in
patients clinically referred to the laboratory as classical MFS and 61/105 (58%) in those referred as incomplete MFS. Conversely, a very low mutation yield 3/21 (14.3%) was observed for
patients referred as possible MFS, but with no major diagnostic criterion, in an organ system. These patients constituted our non-MFS group. The number of mutations detected depending on the
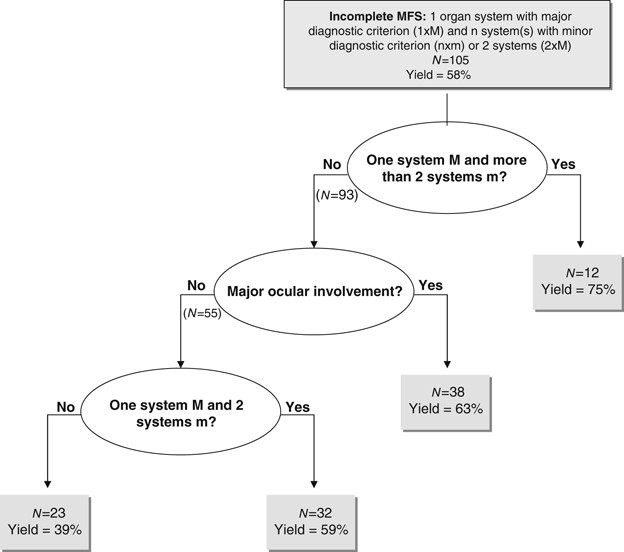
nature of an isolated affected system and the involvement of various system combinations was investigated (Table 1). Among the incomplete MFS patients in whom the _FBN1_ gene had been
screened, we looked for an association between combinations of system involvement and mutation identification rates. For this purpose we performed recursive partitioning, which produced the
regression tree shown in Figure 1. Only probands with available (positive or negative) data for all systems (but neurological) were used. This analysis showed that among patients with
incomplete MFS, the most important criteria for mutation detection were the number of systems involved and the presence of EL. MUTATION IN OTHER DISEASES _FBN1_ gene mutations were found in
a few probands carrying overlapping diseases: c.3058A>G in a Lujan Fryns patient, c.2088C>A and c.4270C>G in two SGS patients, c.3761G>A in the only FS-LDS1A patient screened and
c.640G>A was detected in one WMS patient. Finally, no mutation was found in the other LDS patients and in EDS patients. DISCUSSION This study represents the largest report of _FBN1_
mutations from a single laboratory and shows that France is one of the leading countries for molecular diagnosis of type 1 fibrillinopathies. In France, until recently, only our laboratory
was implicated in _FBN1_ mutation screening. We chose to apply systematic sequencing of the 65 exons of the _FBN1_ gene for detection and identification of mutations. This strategy was
performed on a series of 586 probands that enabled us to identify 354 mutations in 345 probands. FRENCH MUTATIONS COMPARED WITH EARLIER REPORTED MUTATIONS The large set of molecular data
that we report generally supports the observations made by various teams on much smaller data sets (Supplementary Table 7). The percentage of missense, splice sites, nonsense mutations,
insertions/duplications, and deletions are roughly comparable with earlier reported mutations.11, 28, 29 A slight decrease in deletion mutations to the advantage of point mutations, and
particularly nonsense mutations, is observed. These mutations are, as earlier described mutations, spread along the _FBN1_ gene without specific clustering, sign of a mutation hotspot
(Supplementary Table 7). An increased number of mutations is found for exons: 2, 15, 22, 27, 46, 55, and 62 and, conversely, an apparent lack of mutations is found in exons 7, 41, and 65. Of
all the _FBN1_ mutations described today (more than 1750, data not published), the great majority (79.1%) is from the European laboratories and especially Western Europe (France, UK,
Germany, Italy, Belgium, Norway, and Netherland in order of importance, according to UMD-FBN1). These higher number of mutations found in Europe is related to the fact that _FBN1_ mutation
screening is no longer performed on a regular basis in academic laboratories elsewhere. It is interesting to note that this report is the first to describe several double mutants. In effect,
nine probands carried two mutations on the same allele. The lack of comparable information in reports from other teams could be explained either by incomplete screening of the gene when a
first mutation is found, or that other teams have in fact only reported the most probably deleterious event. In our study, we found a significant difference in the yield of _FBN1_ mutation
identification between patients fulfilling and not fulfilling Ghent diagnostic criteria (72.5 _vs_ 58% _P_=0.0001). This is in agreement with earlier reports that are summarized along with
ours in Table 2. More mutations were found in our study ‘incomplete MFS group’ (58%) than in other studies. This may be explained by the fact that our incomplete MFS population is not
comparable with those reported by other teams.28, 29, 30, 31 The incomplete MFS groups in the latter are in fact a combination of our incomplete MFS and our non-MFS groups. Furthermore, it
should be noted that in all these published studies, the authors report some patients in whom the status of the dura is unknown, thus comparable with our report.4, 28, 29, 30, 31 Taken
together results from all studies confirm our first published recommendation:4 that _FBN1_ gene screening be performed in newly suspected MFS when two systems are involved with at least one
major diagnostic criterion. IDENTIFICATION OF INDICATORS FOR BETTER MUTATION DETECTION RATES To understand the correlation between organ system involvement and mutation finding, we checked
the contribution of each system alone or in various combinations. Taken singly, each organ system, even with a major diagnostic criterion, was not by itself a good predictor of mutation
identification (Table 1). However, major ocular involvement in various combinations was always associated with an elevated yield in mutation identification. In the case of major cardiac
involvement, an interesting trend was found: mutation detection rate increased by steps for each added minor system involvement. Recursive partition allowed us to assess the relative
importance for mutation identification of the type of system involvement and the number of systems involved with a minor diagnostic criterion. This data-mining technique naturally takes into
account interaction between variables and produces easy-to-use regression trees. Among the different variables tested, we found that the most significant was the number of organ systems
involved. Thus in our study, the best predictor of the identification of a mutation in the _FBN1_ gene was the presence of features in a wide number of systems. This result further
emphasizes the importance of an exhaustive clinical investigation to evaluate the pertinence of performing the molecular analysis. The second most significant variable found in our study was
EL. Therefore, the second best predictor of identification of a mutation in the _FBN1_ gene was the combination of EL and involvement of at least another system. The importance of EL with
respect to probability of mutation identification has already been noted.32 However, our study shows that this major diagnostic criterion alone is insufficient. RECOMMENDATIONS IN CHOICE OF
PATIENTS TO BE SCREENED Laboratories offering molecular diagnosis for MFS and overlapping disorders must meet a challenge to provide diagnosis in the most relevant clinical situations, while
managing costs. Since the identification of the first mutations in the _FBN_1 gene in MFS patients, laboratories have been flooded with prescriptions for molecular screening of the gene in
probands distributed throughout a wide clinical spectrum ranging from neonatal MFS to isolated skeletal overgrowth. Rapidly, most diagnostic laboratories generally performed _FBN1_ gene
screening in three circumstances: first, in probands meeting the clinical criteria for MFS to offer diagnosis for at-risk relatives; second, in children highly suspected of MFS, but failing
to meet the diagnostic criteria, as features of the disease appear over time (from childhood to early adult); and third when prenatal diagnosis is considered for an at-risk couple. In the
first two circumstances, molecular diagnosis enables proper and early identification of affected patients who need adequate follow-up and treatment to prevent the life-threatening
complications of MFS. However, these circumstances exclude a large body of probands with an increased risk of aortic dilatation and dissection because they carry an _FBN1_ mutation.
Therefore, we not only confirm that _FBN1_ gene screening should be performed in newly suspected MFS when two systems are involved with at least one major diagnostic criterion, but we also
show that within this context, the yield of mutation identification will be the highest in probands presenting with EL and involvement of at least another system. Finally, our data also show
that our original recommendation (two systems involved with at least one with major criterion)4 represent the minimal criteria, because in probands not meeting these criteria, the yield of
mutation identification drastically falls. REFERENCES * Ammash NM, Sundt TM, Connolly HM : Marfan syndrome-diagnosis and management. _Curr Probl Cardiol_ 2008; 33: 7–39. Article Google
Scholar * Judge DP, Dietz HC : Marfan's syndrome. _Lancet_ 2005; 366: 1965–1976. Article CAS Google Scholar * Boileau C, Jondeau G, Mizuguchi T, Matsumoto N : Molecular genetics of
Marfan syndrome. _Curr Opin Cardiol_ 2005; 20: 194–200. Article Google Scholar * Faivre L, Collod-Beroud G, Loeys BL _et al_: Contribution of molecular analyses in diagnosing Marfan
syndrome and type I fibrillinopathies: an international study of 1009 probands. _J Med Genet_ 2008; 45: 384–390. Article CAS Google Scholar * Corson GM, Chalberg SC, Dietz HC, Charbonneau
NL, Sakai LY : Fibrillin binds calcium and is coded by cDNAs that reveal a multidomain structure and alternatively spliced exons at the 5′ end. _Genomics_ 1993; 17: 476–484. Article CAS
Google Scholar * Pereira L, D’Alessio M, Ramirez F _et al_: Genomic organization of the sequence coding for fibrillin, the defective gene product in Marfan syndrome. _Hum Mol Genet_ 1993;
2: 1762. Article CAS Google Scholar * Kainulainen K, Karttunen L, Puhakka L, Sakai L, Peltonen L : Mutations in the fibrillin gene responsible for dominant ectopia lentis and neonatal
Marfan syndrome. _Nat Genet_ 1994; 6: 64–69. Article CAS Google Scholar * Nijbroek G, Sood S, McIntosh I _et al_: Fifteen novel FBN1 mutations causing Marfan syndrome detected by
heteroduplex analysis of genomic amplicons. _Am J Hum Genet_ 1995; 57: 8–21. CAS PubMed PubMed Central Google Scholar * Putnam EA, Cho M, Zinn AB, Towbin JA, Byers PH, Milewicz DM :
Delineation of the Marfan phenotype associated with mutations in exons 23-32 of the FBN1 gene. _Am J Med Genet_ 1996; 62: 233–242. Article CAS Google Scholar * Wang M, Price C, Han J _et
al_: Recurrent mis-splicing of fibrillin exon 32 in two patients with neonatal Marfan syndrome. _Hum Mol Genet_ 1995; 4: 607–613. Article CAS Google Scholar * Faivre L, Collod-Beroud G,
Loeys BL _et al_: Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. _Am J Hum
Genet_ 2007; 81: 454–466. Article CAS Google Scholar * De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE : Revised diagnostic criteria for the Marfan syndrome. _Am J Med Genet_
1996; 62: 417–426. Article CAS Google Scholar * Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ : Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997.
Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). _Am J Med Genet_ 1998; 77: 31–37. Article CAS Google Scholar * Faivre L, Dollfus H, Lyonnet S _et al_:
Clinical homogeneity and genetic heterogeneity in Weill-Marchesani syndrome. _Am J Med Genet A_ 2003; 123: 204–207. Article Google Scholar * Fryns JP, Buttiens M : X-linked mental
retardation with marfanoid habitus. _Am J Med Genet_ 1987; 28: 267–274. Article CAS Google Scholar * Greally MT, Carey JC, Milewicz DM _et al_: Shprintzen-Goldberg syndrome: a clinical
analysis. _Am J Med Genet_ 1998; 76: 202–212. Article CAS Google Scholar * Loeys BL, Chen J, Neptune ER _et al_: A syndrome of altered cardiovascular, craniofacial, neurocognitive and
skeletal development caused by mutations in TGFBR1 or TGFBR2. _Nat Genet_ 2005; 37: 275–281. Article CAS Google Scholar * Furlong J, Kurczynski TW, Hennessy JR : New Marfanoid syndrome
with craniosynostosis. _Am J Med Genet_ 1987; 26: 599–604. Article CAS Google Scholar * Collod-Beroud G, Lackmy-Port-Lys M, Jondeau G _et al_: Demonstration of the recurrence of
Marfan-like skeletal and cardiovascular manifestations due to germline mosaicism for an FBN1 mutation. _Am J Hum Genet_ 1999; 65: 917–921. Article CAS Google Scholar * Shinawi M, Boileau
C, Brik R, Mandel H, Bentur L : Splicing mutation in the fibrillin-1 gene associated with neonatal Marfan syndrome and severe pulmonary emphysema with tracheobronchomalacia. _Pediatr
Pulmonol_ 2005; 39: 374–378. Article Google Scholar * Collod-Beroud G, Le Bourdelles S, Ades L _et al_: Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism
database. _Hum Mutat_ 2003; 22: 199–208. Article CAS Google Scholar * Beroud C, Hamroun D, Collod-Beroud G, Boileau C, Soussi T, Claustres M : UMD (universal mutation database): 2005
update. _Hum Mutat_ 2005; 26: 184–191. Article CAS Google Scholar * Collod G, Beroud C, Soussi T, Junien C, Boileau C : Software and database for the analysis of mutations in the human
FBN1 gene. _Nucleic Acids Res_ 1996; 24: 137–140. Article CAS Google Scholar * Liu WO, Oefner PJ, Qian C, Odom RS, Francke U : Denaturing HPLC-identified novel FBN1 mutations,
polymorphisms, and sequence variants in Marfan syndrome and related connective tissue disorders. _Genet Test_ 1997; 1: 237–242. Article CAS Google Scholar * Wang M, Kishnani P,
Decker-Phillips M, Kahler SG, Chen YT, Godfrey M : Double mutant fibrillin-1 (FBN1) allele in a patient with neonatal Marfan syndrome. _J Med Genet_ 1996; 33: 760–763. Article CAS Google
Scholar * Downing AK, Knott V, Werner JM, Cardy CM, Campbell ID, Handford PA : Solution structure of a pair of calcium-binding epidermal growth factor-like domains: implications for the
Marfan syndrome and other genetic disorders. _Cell_ 1996; 85: 597–605. Article CAS Google Scholar * Dietz HC, Pyeritz RE : Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan
syndrome and related disorders. _Hum Mol Genet_ 1995; 4 (Spec No): 1799–1809. Article CAS Google Scholar * Loeys B, Nuytinck L, Delvaux I, De Bie S, De Paepe A : Genotype and phenotype
analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. _Arch Intern Med_ 2001; 161: 2447–2454. Article CAS Google Scholar
* Comeglio P, Johnson P, Arno G _et al_: The importance of mutation detection in Marfan syndrome and Marfan-related disorders: report of 193 FBN1 mutations. _Hum Mutat_ 2007; 28: 928.
Article Google Scholar * Biggin A, Holman K, Brett M, Bennetts B, Ades L : Detection of thirty novel FBN1 mutations in patients with Marfan syndrome or a related fibrillinopathy. _Hum
Mutat_ 2004; 23: 99. Article Google Scholar * Rommel K, Karck M, Haverich A _et al_: Identification of 29 novel and nine recurrent fibrillin-1 (FBN1) mutations and genotype-phenotype
correlations in 76 patients with Marfan syndrome. _Hum Mutat_ 2005; 26: 529–539. Article CAS Google Scholar * Loeys B, De Backer J, Van Acker P _et al_: Comprehensive molecular screening
of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. _Hum Mutat_ 2004; 24: 140–146. Article CAS Google Scholar * Hayward C, Porteous ME, Brock DJ : Mutation screening
of all 65 exons of the fibrillin-1 gene in 60 patients with Marfan syndrome: report of 12 novel mutations. _Hum Mutat_ 1997; 10: 280–289. Article CAS Google Scholar * Matsukawa R, Iida K,
Nakayama M _et al_: Eight novel mutations of the FBN1 gene found in Japanese patients with Marfan syndrome. _Hum Mutat_ 2001; 17: 71–72. Article CAS Google Scholar * Halliday DJ,
Hutchinson S, Lonie L _et al_: Twelve novel FBN1 mutations in Marfan syndrome and Marfan related phenotypes test the feasibility of FBN1 mutation testing in clinical practice. _J Med Genet_
2002; 39: 589–593. Article CAS Google Scholar * Katzke S, Booms P, Tiecke F _et al_: TGGE screening of the entire FBN1 coding sequence in 126 individuals with Marfan syndrome and related
fibrillinopathies. _Hum Mutat_ 2002; 20: 197–208. Article CAS Google Scholar * Korkko J, Kaitila I, Lonnqvist L, Peltonen L, Ala-Kokko L : Sensitivity of conformation sensitive gel
electrophoresis in detecting mutations in Marfan syndrome and related conditions. _J Med Genet_ 2002; 39: 34–41. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank
all physicians who referred patients for diagnosis and notably: H Plauchu (Lyon), Clarisse Baumann (Paris), Marlène Rio (Paris), Stanislas Lyonnet (Paris), and Didier Lacombe (Bordeaux).
This study was supported in part by grants from GIS-maladies rares 2004; ANR-05-PCOD-014, Université Versailles Saint Quentin (Legs de Melle Bonnevie); Assistance Publique Hôpitaux de Paris
(CIRC 2007); French Society of Cardiology and French Federation of Cardiology.33, 34, 35, 36, 37 AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * AP-HP, Hôpital Ambroise Paré, Service de
Pédiatrie, Boulogne, France Chantal Stheneur & Bertrand Chevallier * AP-HP, Hôpital Bichat, Consultation Multidisciplinaire Marfan, Paris, France Chantal Stheneur, Laurent Gouya,
Jean-Marie Le Parc, Bertrand Moura, Christine Muti, Bernard Grandchamp, Gilles Sultan, Bertrand Chevallier, Guillaume Jondeau & Catherine Boileau * INSERM, U747, Paris, France Chantal
Stheneur * INSERM, U827, Montpellier, France Gwenaëlle Collod-Béroud & Mireille Claustres * Université Montpellier1, Montpellier, France Gwenaëlle Collod-Béroud & Mireille Claustres
* CHRU Dijon, Centre d’Investigation Clinique–Epidémiologie Clinique/Essais Cliniques, Dijon, France Laurence Faivre * CHRU Dijon, Centre de Génétique, Dijon, France Laurence Faivre * AP-HP,
Hôpital Ambroise Paré, Unité de Recherche Clinique, Boulogne, France Jean François Buyck & Philippe Aegerter * AP-HP, Hôpital Ambroise Paré, Laboratoire Central de Biochimie
d’Hormonologie et de Génétique moléculaire, Boulogne, France Laurent Gouya, Christine Muti & Catherine Boileau * INSERM, U793, Paris, France Laurent Gouya & Bernard Grandchamp *
Université Versailles-SQY, Versailles, France Laurent Gouya, Jean-Marie Le Parc, Philippe Aegerter, Bertrand Chevallier & Catherine Boileau * AP-HP, Hôpital Ambroise Paré, Service de
Rhumatologie, Boulogne, France Jean-Marie Le Parc & Bertrand Moura * AP-HP, Hôpital Bichat, Service de Biochimie Hormonale et Génétique, Paris, France Bernard Grandchamp * AP-HP, Hôpital
Ambroise Paré, Service d’Ophtalmologie, Boulogne, France Gilles Sultan * CHU Montpellier, Laboratoire de Génétique Moléculaire, Montpellier, France Mireille Claustres * AP-HP, Hôpital
Bichat, Service de Cardiologie, Paris, France Guillaume Jondeau * INSERM, U698, Paris, France Guillaume Jondeau * INSERM, U781, Paris, France Catherine Boileau Authors * Chantal Stheneur
View author publications You can also search for this author inPubMed Google Scholar * Gwenaëlle Collod-Béroud View author publications You can also search for this author inPubMed Google
Scholar * Laurence Faivre View author publications You can also search for this author inPubMed Google Scholar * Jean François Buyck View author publications You can also search for this
author inPubMed Google Scholar * Laurent Gouya View author publications You can also search for this author inPubMed Google Scholar * Jean-Marie Le Parc View author publications You can also
search for this author inPubMed Google Scholar * Bertrand Moura View author publications You can also search for this author inPubMed Google Scholar * Christine Muti View author
publications You can also search for this author inPubMed Google Scholar * Bernard Grandchamp View author publications You can also search for this author inPubMed Google Scholar * Gilles
Sultan View author publications You can also search for this author inPubMed Google Scholar * Mireille Claustres View author publications You can also search for this author inPubMed Google
Scholar * Philippe Aegerter View author publications You can also search for this author inPubMed Google Scholar * Bertrand Chevallier View author publications You can also search for this
author inPubMed Google Scholar * Guillaume Jondeau View author publications You can also search for this author inPubMed Google Scholar * Catherine Boileau View author publications You can
also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Chantal Stheneur. ADDITIONAL INFORMATION Supplementary Information accompanies the paper on
European Journal of Human Genetics website (http://www.nature.com/ejhg) SUPPLEMENTARY INFORMATION SUPPLEMENTARY TABLE 1 (DOC 453 KB) SUPPLEMENTARY TABLES 2–7 (DOC 3321 KB) RIGHTS AND
PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Stheneur, C., Collod-Béroud, G., Faivre, L. _et al._ Identification of the minimal combination of clinical features
in probands for efficient mutation detection in the _FBN1_ gene. _Eur J Hum Genet_ 17, 1121–1128 (2009). https://doi.org/10.1038/ejhg.2009.36 Download citation * Received: 29 August 2008 *
Revised: 10 February 2009 * Accepted: 11 February 2009 * Published: 18 March 2009 * Issue Date: September 2009 * DOI: https://doi.org/10.1038/ejhg.2009.36 SHARE THIS ARTICLE Anyone you share
the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative KEYWORDS * Marfan syndrome * fibrillin-1 * mutation analysis
Trending News
Clint Eastwood once branded ‘intimidating as hell’ by Tom Hanks: ‘Treats us like horses!''Where Eagles Dare', starring Clint Eastwood, airs on ITV 4 today. The World War 2 action film, written by Alistair MacL...
Mark bittman’s summer recipes for two | members onlyThere is a food-related reason people love the Riviera, which can be taken to mean Provence, France, and Liguria, Italy...
Guidance updated to support the safe management of funeralsNews story GUIDANCE UPDATED TO SUPPORT THE SAFE MANAGEMENT OF FUNERALS Further guidance for those managing or organising...
A pocket guide to your money and personal finance at age 60 - aarp...2. FIND BALANCE INVESTING IN YOU At least once a year you should rebalance your holdings so that you have the appropriat...
Tripura: tugp alleges huge crisis of food in tribal areasTripura Upajati Ganamukti Parishad on Saturday submitted a letter to the chief minister alleging huge crisis of food and...
Latests News
Identification of the minimal combination of clinical features in probands for efficient mutation detection in the fbn1 geneABSTRACT Mutations identified in the fibrillin-1 (_FBN1_) gene have been associated with Marfan syndrome (MFS). Molecula...
Blaming ministers for every failure drives the centralisation of power | thearticleThe problems which the government has faced with Public Health England and with Ofqual show how difficult it is to disco...
Llm1095 - introduction to lloyd's: basic concepts and terms: managing agents and premium trust funds: aligned member syndicates - hmrc internal manualLLM1095 - INTRODUCTION TO LLOYD'S: BASIC CONCEPTS AND TERMS: MANAGING AGENTS AND PREMIUM TRUST FUNDS: ALIGNED MEMBE...
Aarp supplier diversity in actionMemorial Day Sale! Join AARP for just $11 per year with a 5-year membership Join now and get a FREE gift. Expires 6/4 G...
Mitochondrial targeting by dichloroacetate improves outcome following hemorrhagic shockABSTRACT Hemorrhagic shock is a leading cause of death in people under the age of 45 and accounts for almost half of tra...