Stability of the cancer target ddias is regulated by the chip/hsp70 pathway in lung cancer cells
Stability of the cancer target ddias is regulated by the chip/hsp70 pathway in lung cancer cells"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT DNA damage-induced apoptosis suppressor (DDIAS) rescues lung cancer cells from apoptosis in response to DNA damage. DDIAS is transcriptionally activated by NFATc1 and EGF-mediated
ERK5/MEF2B, leading to cisplatin resistance and cell invasion. Therefore, DDIAS is suggested as a therapeutic target for lung cancer. Here, we report that DDIAS stability is regulated by E3
U-box ubiquitin ligase carboxyl terminus of HSP70-interacting protein (CHIP)-mediated proteasomal degradation. We first isolated CHIP as an interacting partner of DDIAS by yeast two-hybrid
screening. CHIP physically associated with both the N- and C-terminal regions of DDIAS, targeting it for proteasomal degradation and reducing the DDIAS half-life. CHIP overexpression
analyses indicated that the tetratrico peptide repeat (TPR) domain and the U-box are required for DDIAS ubiquitination. It is likely that HSP70-bound DDIAS is recruited to the CHIP E3 ligase
via the TPR domain, suggesting DDIAS as a client protein of HSP70. In addition, CHIP overexpression in lung cancer cells expressing high DDIAS levels induced significant growth inhibition
by enhancing DDIAS degradation. Furthermore, simultaneous CHIP overexpression and DNA damage agent treatment caused a substantial increase in the apoptosis of lung cancer cells. Taken
together, these findings indicate that the stability of the DDIAS protein is regulated by CHIP/HSP70-mediated proteasomal degradation and that CHIP overexpression stimulates the apoptosis of
lung cancer cells in response to DNA-damaging agents. SIMILAR CONTENT BEING VIEWED BY OTHERS STAMBPL1 PROMOTES BREAST CANCER CELL RESISTANCE TO CISPLATIN PARTIALLY BY STABILIZING MKP-1
EXPRESSION Article 02 March 2022 DNAJB9 SUPPRESSES THE METASTASIS OF TRIPLE-NEGATIVE BREAST CANCER BY PROMOTING FBXO45-MEDIATED DEGRADATION OF ZEB1 Article Open access 08 May 2021 CONTROL OF
SOX2 PROTEIN STABILITY AND TUMORIGENIC ACTIVITY BY E3 LIGASE CHIP IN ESOPHAGEAL CANCER CELLS Article 23 June 2023 MAIN Protein turnover is an essential process in regulating cellular
function, which prevents the accumulation of unnecessary proteins. E3 U-box ubiquitin ligase carboxyl terminus of HSP70-interacting protein (CHIP) has crucial roles in maintaining the
steady-state levels of a number of target proteins via the ubiquitin–proteasome system.1, 2, 3, 4 CHIP contains a U-box for interaction with the E2 ubiquitin conjugating enzyme and directs
the ubiquitination of substrates and a tetratrico peptide repeat region (TPR) domain for interaction with the chaperones heat shock protein 70 (HSP70) or HSP90. CHIP is involved not only in
homeostatic regulation under resting conditions but also in various stress-activated signaling pathways.4, 5 Therefore, CHIP is associated with diseases such as cancer, neurological
disorders, cardiac disease and bone metabolism.4 CHIP has been considered as a tumor suppressor because it negatively regulates oncoproteins such as Akt, hypoxia inducible factor 1_α_
(HIF-1_α_), BCR-ABL, human telomerase reverse transcriptase (hTERT) and c-Myc.6, 7, 8 In accordance with this, CHIP expression is significantly downregulated in a number of cancers.9 In
contrast, CHIP also functions as an oncogene by targeting phosphatase and tensin homolog (PTEN) for proteasomal degradation in prostate cancer cells.10 CHIP-null mice are
temperature-sensitive and develop multi-organ apoptosis after heat shock, indicating the role of CHIP in the response to stress.11 In addition, CHIP suppresses cancer stem cell properties in
breast cancer cells.12 CHIP is also involved in the degradation of proteins during single-strand break repair/base excision repair.13, 14, 15 Recently, DNA damage-induced apoptosis
suppressor (DDIAS) has been suggested as a promising therapeutic target in several cancers.16, 17, 18, 19 DDIAS is highly expressed in lung cancer and hepatocellular carcinoma cells and
promotes cellular proliferation, colony formation, cellular migration and _in vivo_ tumorigenicity.16, 17, 18 We have previously revealed that DDIAS knockdown induces apoptosis in cancer but
not normal cells.16 In addition, we demonstrated that DDIAS knockdown concomitant with exposure to DNA-damaging agents enhances cancer cell death synergistically. In contrast, DDIAS
overexpression restores the induction of lung cancer cell apoptosis by DNA damage agents, indicating that DDIAS functions as a DNA damage-induced suppressor of apoptosis.16, 19 DDIAS is
transcriptionally activated by nuclear factor of activated T cells (NFAT2).18, 19 In response to epidermal growth factor (EGF), the transcription of DDIAS is also activated via ERK5/MEF2B
signaling, thereby promoting cell invasion by _β_-catenin accumulation.20 In addition to inhibiting DDIAS transcription, targeting DDIAS degradation mechanisms might also be crucial to
suppress cancer cell growth. However, little is known regarding the DDIAS protein degradation process. Therefore, we investigated the mechanism that regulates DDIAS protein turnover. Here,
we report that DDIAS protein turnover is regulated by CHIP-mediated proteasomal degradation. We also demonstrate that CHIP overexpression is linked to the growth inhibition of lung cancer
cells by enhancing DDIAS degradation. RESULTS IDENTIFICATION OF CHIP AS A REGULATOR OF DDIAS PROTEASOMAL DEGRADATION To search for proteins that interacted with DDIAS, we performed yeast
two-hybrid screening. The fusion protein of the DNA-binding domain in the GAL4 transcription factor with the C-terminal region of DDIAS (aa 784–998) was used as bait because the remaining
DDIAS protein sequence exhibited false positives owing to DNA-binding activity (Supplementary Figure S1). A HeLa cell cDNA library fused with the activation domain of GAL4 was used as the
prey. From this analysis, we identified 23 genes encoding potential binding partners for DDIAS including _CHIP_ (_STUB1_, NM_005861), _EFEMP1_ (NM_001039349), _ECM1_ (NM_001202858), _FBLN1_
(NM_001996/NM_006486), _CBY1_ (NM_001002880), _MIF4GD_ (NM_020679), _ACTN4_ (NM_004924), _ACTN1_ (NM_001130005), _MAF1_ (NM_032272), _PLSCR1_ (NM_021105) and _ACLY_ (NM_198830). To
investigate the mechanism of DDIAS turnover in cells, we selected the E3 ubiquitin ligase CHIP for further study because CHIP promotes the ubiquitin ligation/chain elongation-mediated
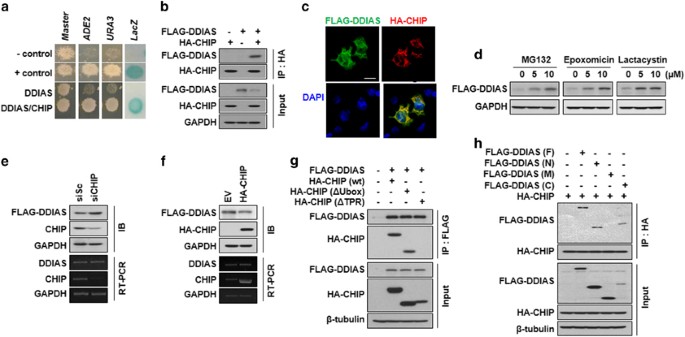
proteasomal degradation of target genes in cancer cells.12, 15, 21 First, we demonstrated that DDIAS interacted with CHIP using three reporters (URA3, lacZ and ADE2) in yeast. Specifically,
the transformant expressing DDIAS (aa 784–998) and CHIP grew well on the plate lacking adenine (SD-ALW) or uracil (SD-ULW), whereas negative control cells expressing only DDIAS or CHIP did
not (Figure 1a). Similarly, the transformant expressing both DDIAS (aa 784–998) and CHIP also exhibited _β_-galactosidase activity, whereas negative controls did not. The dimerization of
polypyrimidine tract-binding protein (PTB) in cells containing both pGBKT7-_PTB_ and pGADT7-_PTB_ served as a positive control. Next, we assessed the physical interaction of DDIAS with CHIP
in mammalian cells. Coimmunoprecipitation (Co-IP) assays clearly showed that FLAG-DDIAS interacted with HA-CHIP _in vivo_ (Figure 1b). Upon immunocytochemical staining, DDIAS co-localized
with CHIP primarily in the cytoplasm of cells (Figure 1c). As CHIP promotes proteasomal degradation through the ligation of ubiquitin to its target proteins, we tested whether DDIAS
undergoes degradation via ubiquitin-mediated proteasomal degradation in HEK293T cells. In the presence of the proteasomal inhibitors MG132, epoxomicin or lactacystin, DDIAS protein levels
greatly increased (Figure 1d), suggesting that DDIAS normally undergoes ubiquitin-mediated proteasomal degradation. We next evaluated whether CHIP was associated with the degradation of
DDIAS. CHIP genetic knockdown significantly increased the FLAG-DDIAS protein level but not the _DDIAS_ mRNA level (Figure 1e). In contrast, HA-CHIP overexpression resulted in a reduction of
the FLAG-DDIAS protein level, whereas again no alteration was observed in the _DDIAS_ mRNA level (Figure 1f). This result suggests that the E3 ligase CHIP interacted with DDIAS in the
cytoplasm, leading to its proteasomal degradation. To determine which domain of CHIP interacted with DDIAS, we utilized Co-IP in HEK293T cells (Figure 1g). No DDIAS binding was observed
following deletion of the CHIP TPR domain, whereas deletion of the U-box demonstrated DDIAS binding. This indicated that the TPR domain was required for the binding of CHIP to DDIAS. We then
examined which domain of FLAG-DDIAS interacted with HA-CHIP _in vivo_ using Co-IP (Figure 1h). The domain analysis of DDIAS demonstrated that polypeptides of both the N (aa 1–400) and C (aa
601–998) termini but not of M (aa 401–600) interacted with the HA-CHIP protein (Figure 1h). Taken together, these results imply that DDIAS turnover is regulated by CHIP-mediated proteasomal
degradation through binding of the TPR domain of CHIP to the N- and C-terminal regions of DDIAS in HEK293T cells. HSP70 RECRUITS DDIAS TO THE CHIP E3 LIGASE, WHEREAS CHIP PROMOTES THE
UBIQUITINATION OF DDIAS CHIP interacts with chaperone HSP70 to affect the ubiquitination of target proteins. Because DDIAS interacted with the TPR domain responsible for HSP70-binding, we
asked whether HSP70 is involved in the degradation of DDIAS as a chaperone in HEK293T cells. FLAG-DDIAS clearly interacted with HSP70 _in vivo_ as shown by a Co-IP assay (Figure 2a). In
addition, HSP70 knockdown significantly increased FLAG-DDIAS protein level without alteration of _DDIAS_ mRNA level (Figure 2b). Conversely, Myc-HSP70 overexpression reduced level of DDIAS,
suggesting that DDIAS degradation is regulated by CHIP in an HSP70-dependent manner (Figure 2c). To investigate the function of HSP70 in the CHIP-mediated proteasomal degradation of DDIAS,
we determined the binding strength between CHIP and DDIAS in the absence of HSP70. Notably, CHIP knockdown did not significantly affect the interaction between DDIAS and HSP70 in the
presence of MG132 (Figure 2d). However, HSP70 knockdown dramatically weakened the interaction between CHIP and FLAG-DDIAS, suggesting that HSP70 mediates the binding of DDIAS to CHIP (Figure
2e). Accordingly, it is likely that HSP70 recruits DDIAS as its client protein for binding to CHIP, which allows CHIP to associate physically with the N (aa 1–400) and C (aa 601–998)
terminal regions in DDIAS. Next, we examined whether the DDIAS protein was polyubiquitinated for proteasomal degradation by CHIP in HEK293T cells. The polyubiquitination of FLAG-DDIAS was
observed in the presence of MG132, whereas it was barely detected in the absence of MG132. Furthermore, DDIAS polyubiquitination significantly increased in the cells expressing HA-CHIP
compared with those not expressing HA-CHIP (Figure 2f). However, a significant ub-signal is still observed even in the absence of HA-CHIP, presumably due to the presence of endogenous CHIP.
This result indicated that CHIP mediates the ubiquitination of DDIAS for proteasomal degradation. We then assessed which domain of CHIP was important for DDIAS ubiquitination (Figure 2g).
Overexpression of CHIP with deletions of the TPR (CHIP-ΔTPR) or the U-box (CHIP-ΔUbox) demonstrated significant decreases in DDIAS polyubiquitination, indicating that both domains are
required for DDIAS ubiquitination (Figure 2g). This result also supports that HSP70-bound DDIAS is recruited to the CHIP E3 ligase via TPR and then ubiquitinated by the CHIP E3 ligase via
the U-box. CHIP OVEREXPRESSION INCREASES DDIAS TURNOVER IN HEK293T CELLS As CHIP is involved in the proteasomal degradation of DDIAS, we assessed the effect of CHIP on the stability of the
DDIAS in HEK293T cells (Figure 3a). CHIP knockdown increased the half-life of FLAG-DDIAS from approximately 3 to >6 h. In contrast, the overexpression of HA-CHIP reduced the half-life of
FLAG-DDIAS to approximately 1.2 h. This result indicates that CHIP has a crucial role in regulating DDIAS turnover in cells. Therefore, we postulated that the binding of CHIP to the
individual DDIAS domains correlated with their respective stabilities. When the stability of each domain was examined, the CHIP binding, N (aa 1–400) and C (aa 601–998) regions were found to
be more unstable compared with the unbound and M (aa 401–600) regions (Figure 3b). Consistent with this, HA-CHIP overexpression significantly reduced the expression of both the N- (aa
1–400) and C- (aa 601–998) terminal regions of the DDIAS protein but not of M (aa 401–600; Figure 3c). It is likely that the high turnover rate of both the N and C domains is associated with
CHIP-mediated degradation. This observation strongly suggests that CHIP increases turnover of the DDIAS protein by targeting both the N- and C-terminal regions. CHIP LEVELS CORRELATE WITH
DDIAS INSTABILITY AND DDIAS DEPLETION-INDUCED GROWTH INHIBITION IN LUNG CANCER CELLS We previously demonstrated that DDIAS is highly expressed in various cancer tissues, although
tissue-specific DDIAS expression in normal tissues is observed.16 To evaluate the relevance of DDIAS to cancerous properties, we determined the level of DDIAS protein in lung cancer cells.
We found that DDIAS expression was relatively high and varied in lung cancer cells, although it was detected at very low levels in lung fibroblast cells, WI-38 and CCD-34Lu cells (Figure
4a). The expression of both HSP70 and HSP90 was increased in lung cancer cells compared with normal lung fibroblast cells. First, we confirmed endogenous DDIAS level is reduced by CHIP
overexpression in NCI-H1299 and NCI-H1703 (Figure 4b). In NCI-H1299 cells expressing relatively low CHIP, endogenous DDIAS was relatively stable with a long half-life of >12 h (Figure
4c). We then tested whether the forced overexpression of HA-CHIP affected the extended DDIAS stability in NCI-H1299 cells HA-CHIP overexpression significantly decreased the turnover of DDIAS
yielding a half-life of approximately 8 h (Figure 4c). On the contrary, half-life of DDIAS in NCI-H1437 expressing high CHIP was about 9 h, which was increased dramatically to more than 12
h by CHIP knockdown (Figure 4d). This result implicates that CHIP expression correlates with DDIAS instability in lung cancer cells. We previously reported that DDIAS downregulation
suppresses the growth of lung cancer cells.16 Therefore, we assessed whether HA-CHIP overexpression affects the growth of NCI-H1299 and NCI-H1703 cells through DDIAS degradation. Our results
indicated that HA-CHIP overexpression induced DDIAS downregulation and the growth inhibition of NCI-H1299 and NCI-H1703 cells (Figure 4e). In contrast, the overexpression of Flag-DDIAS
partially protected NCI-H1299 and NCI-H1703 cells from the growth inhibition by HA-CHIP overexpression (Figure 4f). We also confirmed that Flag-DDIAS was still present even with HA-CHIP
overexpression (Figure 4g). These findings suggest that CHIP overexpression in lung cancer cells induces growth inhibition by enhancing DDIAS degradation. CHIP OVEREXPRESSION PLUS DNA DAMAGE
SUBSTANTIALLY ENHANCES THE GROWTH INHIBITION OF CANCER CELLS In an earlier study, we demonstrated that DDIAS knockdown induced a synergistic effect on growth inhibition with concomitant
treatment by a DNA damage-inducing agent such as CPT and cisplatin.16 Therefore, we tested whether CHIP overexpression enhances the growth inhibition of NCI-H1299 and NCI-H1703 cells in the
presence of CPT or of perifosine, which acts to induce apoptosis through Akt inhibition, as a negative control (Figure 5a). The growth of HEK293T cells was not significantly affected by
either CPT or perifosine at the concentrations used. HA-CHIP overexpression also had no effect on the growth of HEK293T cells. However, HA-CHIP overexpression with simultaneous CPT treatment
substantially augmented the growth inhibitory effect on NCI-H1703 and NCI-H1299 (Figure 5a). In contrast, perifosine did not significantly affect the growth inhibition caused by CHIP
overexpression in the cells with low endogenous CHIP levels. Previously, we also suggested that DDIAS function is associated with DNA repair in the presence of DNA-damaging agents.
Specifically, simultaneous DDIAS depletion and treatment with a DNA damage-inducing agent caused synergistic effects on DNA damage and the growth inhibition of A549 cells.16 Therefore, we
asked whether HA-CHIP overexpression markedly impacts DNA damage. In comet assays, CPT treatment of cells overexpressing HA-CHIP showed a synergistic effect on DNA damage with longer-tailed
comets in NCI-H1299 and NCI-H1703 cells (Figure 5b). However, concomitant treatment of these cells to both perifosine and CHIP overexpression did not yield this effect. Furthermore, HEK293T
cells showed no detectable change in comet lengths. In addition, western blot analysis also revealed that HA-CHIP overexpression caused a significant effect on the apoptosis of both
NCI-H1299 and NCI-H1703 cells upon CPT exposure, resulting in the increase of PARP-1 cleavage and γH2AX in these cells (Figure 5c). This result suggests that DDIAS depletion mediated by CHIP
overexpression and the simultaneous induction of DNA damage caused significant cell death only in cancer cells expressing high levels of DDIAS. DISCUSSION To examine the mechanism
underlying proteasomal degradation of DDIAS, we first performed a yeast two-hybrid assay and isolated E3 ligase CHIP as a potential DDIAS-interacting partner. Additional DDIAS-interacting
proteins identified included EFEMP1,22 ECM1,23 CBY1,24 PLSCR1 (ref. 25) and MIF4GD,26 which are involved in cell survival, proliferation, metastasis, invasion, resistance to apoptosis and
cell adhesion. We next revealed that CHIP knockdown decreased the turnover rate of exogenous DDIAS, increasing its stability in HEK293T cells. Conversely, CHIP overexpression significantly
increased DDIAS turnover, decreasing its stability. CHIP targeted the N- and C-terminal regions of DDIAS for its ubiquitination and proteasomal degradation. We also demonstrated that HSP70
recruited DDIAS for binding to CHIP, identifying DDIAS as a client protein of HSP70. Therefore, we suggest that DDIAS stability is regulated as a novel target of the CHIP/HSP70-mediated
ubiquitin–proteasomal degradation pathway. As CHIP is associated with chaperones HSP70 or HSP90 for the ubiquitination of target proteins,27 we examined the involvement of HSP90 in the
ubiquitination-mediated degradation of DDIAS. Notably, HSP90 knockdown led to reduced DDIAS protein level (Supplementary Figure S2), suggesting that HSP90 might not function as a chaperone
for DDIAS ubiquitination. However, the findings clearly demonstrated that HSP70 was solely involved in CHIP-mediated proteasomal degradation of DDIAS. DDIAS expression levels differ between
human tissues, and DDIAS knockdown does not noticeably affect the growth of either WI-38 or HEK293T cells.16, 19 Furthermore, we found that HA-CHIP overexpression did not cause either DNA
damage or growth inhibition in HEK293T cells, suggesting that DDIAS downregulation may not seriously compromise normal cells. In addition, we revealed that CHIP expression is lower in
NCI-H1299 and NCI-H1703 cells, in which endogenous DDIAS is more stable with >12 h half-life, compared with the 9 h half-life of DDIAS in NCI-H1437 cells exhibiting high CHIP expression.
However, CHIP overexpression in NCI-H1299 led to increased turnover of DDIAS, resulting in a reduction of DDIAS half-life from >12 to 8 h. In contrast, CHIP knockdown in NCI-H1437
resulted in increased half-life from 9 to >12 h, indicating correlation of CHIP expression level with DDIAS turnover rate. Furthermore, we have shown that CHIP overexpression induced
significant growth inhibition of NCI-H1299 and NCI-H1703 cells through DDIAS downregulation. Thus, we suggest that cells expressing low CHIP level are associated with DDIAS-dependent growth
inhibition and are likely sensitive to DDIAS depletion, as shown for NCI-H1299 and NCI-H1703 cells. In addition, DDIAS overexpression suppressed growth inhibition caused by CHIP
overexpression in NCI-H1299 or NCI-H1703 cells, suggesting CHIP-dependent DDIAS instability is directly linked with the growth of cancer cells. Previously, we showed that simultaneous DDIAS
depletion and DNA damage has synergistic effects on DNA damage and growth inhibition in A549 cells.16 Consistent with this, HA-CHIP overexpression with simultaneous CPT treatment resulted in
a substantial enhancing effect on the induction of apoptosis in NCI-H1703 and NCI-H1299. CHIP overexpression greatly sensitized the former cells to CPT, resulting in a significant
inhibition of cell growth. Furthermore, CPT treatment simultaneous with HA-CHIP overexpression substantially impacted the degree of DNA damage yielding longer-tailed comets in comet assays
in NCI-H1299 and NCI-H1703 cells, whereas this effect was not observed following simultaneous perifosine treatment. Notably, DDIAS is also associated with cisplatin resistance in lung cancer
cells.19 Therefore, it is likely that DDIAS is associated with DNA repair by suppressing DNA damage in the presence of DNA-damaging agents in NCI-H1299 and NCI-H1703 cells. Studies are
currently underway to better understand the involvement of DDIAS in DNA repair. We observed the significant growth recovery by forced DIAAS overexpression in CHIP overexpressing lung cancer
cells, suggesting that increase of DDIAS turnover by CHIP overexpression noticeably contributes to growth inhibition of cells. Recently, many reports have demonstrated that CHIP negatively
regulates the expression of various genes including _VEGFR2_, _c-Myc_ and _PRMT5_, which mediates proliferation, cell migration, invasion and angiogenesis.6, 9, 28 Therefore, we cannot
exclude the possibility of pleiotropic effects of HA-CHIP overexpression on growth inhibition of lung cancer cells, which might represent a limitation of this study. Notably, _DDIAS_ mRNA
expression correlated approximately 73% with NFATc1 protein expression, indicating a crucial role of the transcriptional regulation of this protein.19 However, the inverse correlation
between DDIAS and CHIP expression implicates CHIP/HSP70-mediated degradation of DDIAS as a crucial regulatory system for DDIAS expression. Although the importance of the role of CHIP in
DDIAS expression in cancer cells remains unclear, CHIP-mediated DDIAS instability might cause significant growth inhibition via DNA damage and apoptosis of cancer cells, but not of normal
cells. In summary, we describe the mechanism controlling DDIAS stability by CHIP/HSP70-mediated proteasomal degradation in lung cancer cells. As a client of HSP70, DDIAS is recruited to the
E3 ligase CHIP for proteasomal degradation of the DDIAS protein. In NCI-H1703 and NCI-H1299 cells, CHIP overexpression leads to a significant enhancement of apoptotic cells in the presence
of DNA-damaging agents. MATERIALS AND METHODS REAGENTS AND ANTIBODIES Tween 20, paraformaldehyde, DAPI (4',6-diamidino-2-phenylindole), sulforhodamine B (SRB), cycloheximide (CHX) were
purchased from Sigma-Aldrich (St. Louis, MO, USA). Camptothecin (CPT) and perifosine were obtained from Calbiochem (Darmstadt, Germany) and AdooQ Bioscience LLC (Irvine, CA, USA),
respectively. MG132, epoxomicin and lactacystin were supplied by Cayman Chemicals (Ann Arbor, MI, USA). Chemicals were dissolved in DMSO or water at 20 mM concentrations. Rabbit polyclonal
antibodies against HA, c-Myc and PARP-1, and mouse monoclonal antibody against luciferase were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal _β_-tubulin
antibody was from Abcam (Cambridge, MA, USA). Mouse monoclonal anti-FLAG (M2) and rabbit polyclonal CHIP antibodies were obtained from Sigma-Aldrich. Rabbit polyclonal GAPDH and _β_-actin
antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies were obtained from AbFrontier (Seoul, Korea). Rabbit polyclonal HSP90 antibody was purchased from Enzo Life
Sciences (Farmingdale, NY, USA). FITC or rhodamine B-conjugated secondary antibody and agarose were obtained from Santa Cruz Biotechnology. Anti-FLAG M2 affinity gel and anti-HA agarose were
obtained from Sigma-Aldrich. CELL CULTURE Human embryonic kidney cells expressing the SV40 large T antigen (HEK293T), lung fibroblast cells (WI-38, CCD-34Lu) and lung carcinoma cells
(NCI-H460, HCC-827, NCI-H1299, NCI-H1703, NCI-H1437) were obtained from the American Type Culture Collection (Manassas, VA, USA) or the KRIBB cell line bank (Daejeon, Korea). Cells were
cultured in RPMI 1640 or Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (Gibco), 100 U/ml penicillin and
100 _μ_g/ml streptomycin (Gibco). The cells were maintained in a humidified incubator at 37 °C with 5% CO2. For chemical treamtment of cells, CPT (10 _μ_M, 24 h), cycloheximide (CHX, 20
_μ_M) and perefosine (10 _μ_M, 24 h) were used. YEAST TWO-HYBRID ANALYSIS Yeast two-hybrid assay was performed as described previously.29 DDIAS (784 aa-end of _DDIAS_, 215 aa) was cloned
into BamHI/PstI sites of the pGBKL vector containing the DNA-binding domain of GAL4 (GAL4DB) and _LEU2_ as a selection marker in yeast. A human HeLa cDNA AD library was used. Yeast PBN204
strain contains three reporters (_URA3_, _lacZ_ and _ADE2_) that are under the control of different _GAL_ promoters. Yeast transformants of the DDIAS bait (aa 784–998) and Human HeLa cDNA AD
library were spread on selection media [SD-leucine, tryptophan, uracil (SD-LWU)] that supports growth of yeasts with bait and prey plasmids, yielding proteins that interact with each other.
We tested protein–protein interactions using beta-galactosidase, ADE2 and URA3 reporters. As the negative control, vectors pGBKT7 and pGADT7 were used. Dimerization of polypyrimidine
tract-binding protein (PTB) was used as the positive control by generating pGBKT7-_PTB_ and pGADT7-_PTB_. REVERSE TRANSCRIPTION POLYMERASE CHAIN REACTION Total RNA was extracted from cells
using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and reverse transcribed into cDNA using the TOPscript cDNA synthesis kit (Enzynomics, Daejeon, Korea) according to the manufacturer’s
instructions. Equal amounts of cDNA were diluted and amplified by PCR. The following primers were used: _DDIAS_ forward, 5′-CAGAAGCCCTATTGTATCTGG-3′; _DDIAS_ reverse,
5′-GTGACCAAGCACTTCGAGTTT-3′; _CHIP_ forward, 5′-CGAATCGCGAAGAAGAAGCG-3′; _CHIP_ reverse, 5′-GGTCAAAATGACCCACACGC-3′; _HSP70_ forward, 5′-GCCTACTTCAACGACTCGCA-3′; _HSP70_ reverse,
5′-AGTCGATGCCCTCAAACAGG-3′; 5′-_GAPDH_ forward, 5′-TCATGACCACAGTCCATGCC-3′; and _GAPDH_ reverse, 5′-TCCACCACCCTGTTGCTGTA-3′. GENE KNOCKDOWN DDIAS and scrambled siRNAs were synthesized by ST
Pharm Co., Ltd (Siheung-si, Korea). The sequences of siRNA were used as follows: _DDIAS_, 5′-CUGUAACCCAGGCAGAUGUdTdT-3′; siScramble, 5′-CCUACGCCACCAAUUUCGUdTdT-3′. Non-Targeting
(D-001206-14), _HSP90AA1_(M-005186-02), _HSPA1A_(M-005168-01) and _CHIP_(M-007201-02) from siGENOME SMARTpool were purchased from Dharmacon Inc. (Lafayette, CO, USA). Transfection was
performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) or DharmaFECT 1 (Lafayette, CO, USA) according to the manufacturer’s instructions. SRB ASSAY Cell cytotoxicity was
determined with the SRB assay as described previously.19 Briefly, the cells were fixed with 4% formaldehyde (Sigma-Aldrich) and stained with SRB solution. The SRB dye bound to the cell
matrix was eluted with elution buffer and quantified at 530 nm on a Molecular Devices EMax system (San Diego, CA, USA). GENE CLONING AND TRANSIENT OVEREXPRESSION IN CELLS _DDIAS_ (1–998
amino acids) and _HSP70_ (1-641 amino acids) cDNA clones were obtained from the Korea Human Gene Bank, Medical Genomics Research center, KRIBB, Korea. _DDIAS_ Full-length F (aa 1–998) and
domain N (aa 1–400), M (aa 401–600) and C (aa 601–998) were amplified by PCR and cloned into the p3xFLAG-CMV vector (Sigma-Aldrich) to create the p3xFLAG-CMV-_DDIAS_. _HSP70_-wt (1-641 amino
acids) clone was inserted into pcDNA3 vector with Myc tag (Invitrogen). The coding region of CHIP was obtained by PCR-amplification of HEK293T cDNA. HA-_CHIP_-wt (1-303 amino acids),
HA-_CHIP_-ΔTPR (128–303 amino acids) and HA-_CHIP_-ΔUbox (1–195 amino acids) were amplified by PCR and inserted into the pcDNA3. Transfections were performed using TurboFect (Thermo
Scientific, Rockford, lL, USA) according to the manufacturer’s instructions. IMMUNOCYTOCHEMISTRY Immunocytochemistry was performed using ibidi _μ_-Slides or _μ_-Dishes (Ibidi, München,
Germany) as described.30 The cells were fixed by 4% paraformaldehyde (Sigma) in PBS, washed three times with PBS and permeabilized with 0.4% Triton X-100 in PBS. After blocking with 2% BSA
in PBS for 30 min, the cells were incubated with appropriate primary antibody at 4 °C overnight. The cells were incubated with FITC or rhodamine B-conjugated secondary antibody and observed
using a confocal laser scanning microscope (LSM510 META, Carl Zeiss, Jena, Germany). COMET ASSAY The Comet assay was performed using an OxiSelect Comet Assay Kit (Cell Biolabs, San Diego,
CA, USA) as described by manufacturer.16 Briefly, cells in agarose slide were lysed, treated with alkaline solution and electrophoresed. The precipitated chromosomal DNA with 70% ethanol
were stained with Vista Green DNA Dye and then observed by fluorescent microscopy (Leica DM IL LED, Leica Microsystems, Wetzlar, Germany). CO-IP AND WESTERN BLOT For immunopreciptation, cell
lysates in RIPA buffer (Millipore, Temecula, CA, USA) were incubated with anti-FLAG M2 affinity gel, anti-HA agarose or anti-c-Myc agarose in IP buffer and centrifuged. Agarose pellet was
boiled in 1 × sample buffer. For western blotting, the proteins in cell lysates were separated by SDS-PAGE and transferred to PVDF membrane (Millipore). The membrane was blotted with primary
and secondary antibodies conjugated with HRP substrate (Millipore). The enhanced chemiluminescent was detected using chemiluminescence (ECL) kit (Millipore). The band intensity was
quantified by AlphaEaseFC software (Alpha Innotech, San Leandro, CA, USA). STATISTICAL ANALYSIS All values were shown as the mean±s.d. of one experiment performed in triplicate. Similar
results were obtained from at least three independent experiments. Data were analyzed using Student’s _t_-test for two group’s comparisons or using the Student–Newman–Keuls (SNK) test for
multiple comparisons to determine statistical significance (*_P_<0.05, **_P_<0.01, ***_P_<0.005). REFERENCES * Kundrat L, Regan L . Balance between folding and degradation for
Hsp90-dependent client proteins: a key role for CHIP. _Biochemistry_ 2010; 49: 7428–7438. Article CAS Google Scholar * Matsumura Y, Sakai J, Skach WR . Endoplasmic reticulum protein
quality control is determined by cooperative interactions between Hsp/c70 protein and the CHIP E3 ligase. _J Biol Chem_ 2013; 288: 31069–31079. Article CAS Google Scholar * Millan IC,
Squillace AL, Gava LM, Ramos CH . The stability of wild-type and deletion mutants of human C-terminus Hsp70-interacting protein (CHIP). _Protein Pept Lett_ 2013; 20: 524–529. Article CAS
Google Scholar * Paul I, Ghosh MK . A CHIPotle in physiology and disease. _Int J Biochem Cell Biol_ 2015; 58: 37–52. Article CAS Google Scholar * Lee JS, Seo TW, Yi JH, Shin KS, Yoo SJ .
CHIP has a protective role against oxidative stress-induced cell death through specific regulation of endonuclease G. _Cell Death Dis_ 2013; 4: e666. Article CAS Google Scholar * Paul I,
Ahmed SF, Bhowmik A, Deb S, Ghosh MK . The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. _Oncogene_ 2013; 32: 1284–1295. Article CAS Google Scholar *
Ferreira JV, Fofo H, Bejarano E, Bento CF, Ramalho JS, Girao H _et al_. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. _Autophagy_ 2013; 9: 1349–1366. Article
CAS Google Scholar * Wang S, Wu X, Zhang J, Chen Y, Xu J, Xia X _et al_. CHIP functions as a novel suppressor of tumour angiogenesis with prognostic significance in human gastric cancer.
_Gut_ 2013; 62: 496–508. Article CAS Google Scholar * Sun C, Li HL, Chen HR, Shi ML, Liu QH, Pan ZQ _et al_. Decreased expression of CHIP leads to increased angiogenesis via VEGF-VEGFR2
pathway and poor prognosis in human renal cell carcinoma. _Sci Rep_ 2015; 5: 9774. Article Google Scholar * Ahmed SF, Deb S, Paul I, Chatterjee A, Mandal T, Chatterjee U _et al_. The
chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. _J Biol Chem_ 2012; 287: 15996–16006. Article CAS Google Scholar *
Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V _et al_. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. _EMBO J_ 2003; 22: 5446–5458. Article CAS
Google Scholar * Tsuchiya M, Nakajima Y, Hirata N, Morishita T, Kishimoto H, Kanda Y _et al_. Ubiquitin ligase CHIP suppresses cancer stem cell properties in a population of breast cancer
cells. _Biochem Biophys Res Commun_ 2014; 452: 928–932. Article CAS Google Scholar * Sobol RW . CHIPping away at base excision repair. _Mol Cell_ 2008; 29: 413–415. Article CAS Google
Scholar * Fang Q, Inanc B, Schamus S, Wang XH, Wei L, Brown AR _et al_. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase beta. _Nat Commun_ 2014; 5: 5513.
Article CAS Google Scholar * Parsons JL, Tait PS, Finch D, Dianova II, Allinson SL, Dianov GL . CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision
repair proteins. _Mol Cell_ 2008; 29: 477–487. Article CAS Google Scholar * Won KJ, Im JY, Yun CO, Chung KS, Kim YJ, Lee JS _et al_. Human Noxin is an anti-apoptotic protein in response
to DNA damage of A549 non-small cell lung carcinoma. _Int J Cancer_ 2014; 134: 2595–2604. Article CAS Google Scholar * Nakaya N, Hemish J, Krasnov P, Kim SY, Stasiv Y, Michurina T _et
al_. noxin, a novel stress-induced gene involved in cell cycle and apoptosis. _Mol Cell Biol_ 2007; 27: 5430–5444. Article CAS Google Scholar * Zhang ZZ, Huang J, Wang YP, Cai B, Han ZG .
NOXIN as a cofactor of DNA polymerase-primase complex could promote hepatocellular carcinoma. _Int J Cancer_ 2015; 137: 765–775. Article CAS Google Scholar * Im JY, Lee KW, Won KJ, Kim
BK, Ban HS, Yoon SH _et al_. DNA damage-induced apoptosis suppressor (DDIAS), a novel target of NFATc1, is associated with cisplatin resistance in lung cancer. _Biochim Biophys Acta_ 2015;
1863: 40–49. Article Google Scholar * Im JY, Yoon SH, Kim BK, Ban HS, Won KJ, Chung KS _et al_. DNA-damage–induced apoptosis suppressor (DDIAS) is upregulated via ERK5/MEF2B signaling and
promotes β-catenin-mediated invasion. _Biochim Biophys Acta_ 2016; 1859: 1449–1458. Article CAS Google Scholar * Covey JM, Jaxel C, Kohn KW, Pommier Y, Protein-linked DNA . strand breaks
induced in mammalian cells by camptothecin, an inhibitor of topoisomerase I. _Cancer Res_ 1989; 49: 5016–5022. CAS PubMed Google Scholar * Chen J, Wei D, Zhao Y, Liu X, Zhang J .
Overexpression of EFEMP1 correlates with tumor progression and poor prognosis in human ovarian carcinoma. _PLoS One_ 2013; 8: e78783. Article Google Scholar * Lee KM, Nam K, Oh S, Lim J,
Kim RK, Shim D _et al_. ECM1 regulates tumor metastasis and CSC-like property through stabilization of beta-catenin. _Oncogene_ 2015; 34: 6055–6065. Article CAS Google Scholar *
Mokhtarzada S, Yu C, Brickenden A, Choy WY . Structural characterization of partially disordered human Chibby: insights into its function in the Wnt-signaling pathway. _Biochemistry_ 2011;
50: 715–726. Article CAS Google Scholar * Huang Y, Zhao Q, Zhou CX, Gu ZM, Li D, Xu HZ _et al_. Antileukemic roles of human phospholipid scramblase 1 gene, evidence from inducible
PLSCR1-expressing leukemic cells. _Oncogene_ 2006; 25: 6618–6627. Article CAS Google Scholar * Wan C, Hou S, Ni R, Lv L, Ding Z, Huang X _et al_. MIF4G domain containing protein regulates
cell cycle and hepatic carcinogenesis by antagonizing CDK2-dependent p27 stability. _Oncogene_ 2015; 34: 237–245. Article CAS Google Scholar * Kundrat L, Regan L . Identification of
residues on Hsp70 and Hsp90 ubiquitinated by the cochaperone CHIP. _J Mol Biol_ 2010; 395: 587–594. Article CAS Google Scholar * Zhang HT, Zeng LF, He QY, Tao WA, Zha ZG, Hu CD . The E3
ubiquitin ligase CHIP mediates ubiquitination and proteasomal degradation of PRMT5. _Biochim Biophys Acta_ 2016; 1863: 335–346. Article CAS Google Scholar * Kim DM, Chung KS, Choi SJ,
Jung YJ, Park SK, Han GH _et al_. RhoB induces apoptosis via direct interaction with TNFAIP1 in HeLa cells. _Int J Cancer_ 2009; 125: 2520–2527. Article CAS Google Scholar * Kim BK, Im
JY, Han G, Lee WJ, Won KJ, Chung KS _et al_. p300 cooperates with c-Jun and PARP-1 at the p300 binding site to activate RhoB transcription in NSC126188-mediated apoptosis. _Biochim Biophys
Acta_ 2014; 1839: 364–373. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by National Research Foundation (NRF) grants (2013R1A2A2A01069026 and
2015M3A9A8032460), Health Technology R&D grant (HI13C2162) and the KRIBB Initiative program of the Korea Research Council of Fundamental Science and Technology. AUTHOR INFORMATION Author
notes * Kyoung-Jae Won and Joo-Young Im: These authors contributed equally to this work. AUTHORS AND AFFILIATIONS * Personalized Genomic Medicine Research Center, KRIBB, Daejeon, 305-806,
Korea Kyoung-Jae Won, Joo-Young Im, Bo-Kyung Kim, Young-Jin Jung & Misun Won * Functional Genomics, University of Science and Technology, Daejeon, 305-701, Korea Kyoung-Jae Won &
Misun Won * Metabolic Regulation Research Center, KRIBB, Daejeon, 305-806, Korea Hyun Seung Ban * Oligo Team, ST Pharm. Co., LTD, Sihwa Industrial Complex 1, Kyunggido, 429-848, Korea Kyeong
Eun Jung Authors * Kyoung-Jae Won View author publications You can also search for this author inPubMed Google Scholar * Joo-Young Im View author publications You can also search for this
author inPubMed Google Scholar * Bo-Kyung Kim View author publications You can also search for this author inPubMed Google Scholar * Hyun Seung Ban View author publications You can also
search for this author inPubMed Google Scholar * Young-Jin Jung View author publications You can also search for this author inPubMed Google Scholar * Kyeong Eun Jung View author
publications You can also search for this author inPubMed Google Scholar * Misun Won View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR
Correspondence to Misun Won. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Edited J Chipuk Supplementary Information
accompanies this paper on Cell Death and Disease website SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE 1 (JPG 200 KB) SUPPLEMENTARY FIGURE 2 (JPG 81 KB) RIGHTS AND PERMISSIONS _Cell Death
and Disease_ is an open-access journal published by _Nature Publishing Group_. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third
party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative
Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Won, KJ., Im, JY., Kim, BK. _et al._ Stability of the cancer target DDIAS is regulated by the CHIP/HSP70 pathway in lung cancer
cells. _Cell Death Dis_ 8, e2554 (2018). https://doi.org/10.1038/cddis.2016.488 Download citation * Received: 13 October 2016 * Revised: 15 December 2016 * Accepted: 19 December 2016 *
Published: 12 January 2017 * Issue Date: January 2018 * DOI: https://doi.org/10.1038/cddis.2016.488 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
Trending News
Dolly in nashville: authenticity that makes room for rhinestonesWhen the renowned radio personality and Grand Ole Opry fixture Bill Cody walked onto the stage at the Ryman Auditorioum ...
The latest developments in dentistryThe British Dental Conference & Dentistry Show (BDCDS) is an excellent opportunity to discover the latest developmen...
Festive blether day 11 — scottish national partyThis year, the safest way to spend the festive period is to stay at home. We know it’s not going to be easy and that man...
Mathematical modelling: the cubic map in theory and practiceAccess through your institution Buy or subscribe This is a preview of subscription content, access via your institution ...
Five things to watch at sunday’s academy awards ceremonyFive things to watch at Sunday’s Academy Awards ceremony | WTVB | 1590 AM · 95.5 FM | The Voice of Branch County Close F...
Latests News
Stability of the cancer target ddias is regulated by the chip/hsp70 pathway in lung cancer cellsABSTRACT DNA damage-induced apoptosis suppressor (DDIAS) rescues lung cancer cells from apoptosis in response to DNA dam...
TP53: the unluckiest of genes?Download PDF Perspective Open access Published: 23 October 2024 TP53: the unluckiest of genes? Andreas C. Joerger ORCID...
Polymer journal - volume 11 issue 4, april 1979Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best expe...
Hard skinned aluminium | NPG Asia MaterialsMicro- and nanoscale structures on aluminium provide a superhydrophobic surface, making it resistant to corrosion. Corro...
Measuring cigarette smoking-induced cortical dopamine release: a [11c]flb-457 pet studyABSTRACT Striatal dopamine (DA) is thought to have a fundamental role in the reinforcing effects of tobacco smoking and ...