Nitric oxide signaling regulates mitochondrial number and function
Nitric oxide signaling regulates mitochondrial number and function"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Obesity affects many millions of children and adults worldwide and poses a major health problem. Weight gain results when energy intake exceeds energy expenditure. Energy can be dissipated
in the form of work or heat. In order to combat obesity and associated disease, including diabetes, heart attack, and stroke, understanding how our body regulates energy balance will be of
fundamental importance. Recent findings have revealed that the brain regulates energy expenditure. Environmental cues, such as cold, exercise, and food-intake signal the sympathetic nervous
system to trigger the release of the hormone, noradrenaline, which in turn innervates brown adipose tissue (BAT) by binding to the _β_-adrenergic receptor (reviewed in Lowell and
Spiegelman1). BAT is the major site of adaptive thermogenesis, which protects the body from cold and controls the response to changes in diet. _β_-adrenergic receptor stimulation then leads
to mitochondrial biogenesis (mitochondrial proliferation and activation). The mitochondrion can be viewed as a cellular furnace where fatty acids and glucose are oxidized, and energy is
stored as ATP or wasted as heat, thus regulating cellular energy balance. The exact pathways that lead to mitochondrial proliferation in response to _β_-adrenergic receptor signaling in BAT
are not entirely understood. Clues have emerged indicating that transcriptional control is implicated in this process.2,3,4,5,6 Now Nisoli _et al._7 have made the intriguing discovery that
the gas nitric oxide (NO) links _β_-adrenergic receptor signaling with mitochondrial biogenesis by increasing the activity of a master transcriptional regulator of the mitochondrial
biogenesis program. Here we will highlight the observations made by Nisoli and colleagues and speculate on their potential implications for the study of energy metabolism, aging, and cell
death. _NO regulates mitochondrial biogenesis_. BAT plays an important role in controlling body temperature and energy expenditure. Differentiation of brown adipocytes is accompanied by
multiplication and functional activation of mitochondria.8 This event coincides with the activation of UCP-1, an uncoupling protein of the mitochondrial inner membrane that discharges the
proton gradient, thus dissociating electron transport from ATP synthesis in the respiratory chain and generating heat in response to cold or energy intake.1,9 The proton leak induced by
UCP-1 activity not only generates heat, but also reduces free radical production, thereby regulating redox balance. Nisoli _et al._7 report, in an article recently published in _Science_,
that NO regulates mitochondrial biogenesis in brown adipocytes and various cell lines. NO is known to have pleiotropic physiological effects depending on the target tissue and cell type
(reviewed in Bredt and Snyder10). For instance, in blood vessels, NO functions as a vasodilator, while in the nervous system, NO acts as a neurotransmitter. However, if produced in excess
and in the appropriate redox state, NO can be neurotoxic. NO also regulates the binding and release of oxygen from hemoglobin, thereby regulating oxygen delivery to tissues.11 NO is produced
by three isoforms of the nitric oxide synthase (NOS) enzyme. How then does NO trigger an increase in mitochondrial biogenesis? Nisoli _et al._7 have found that _β_-adrenergic receptor
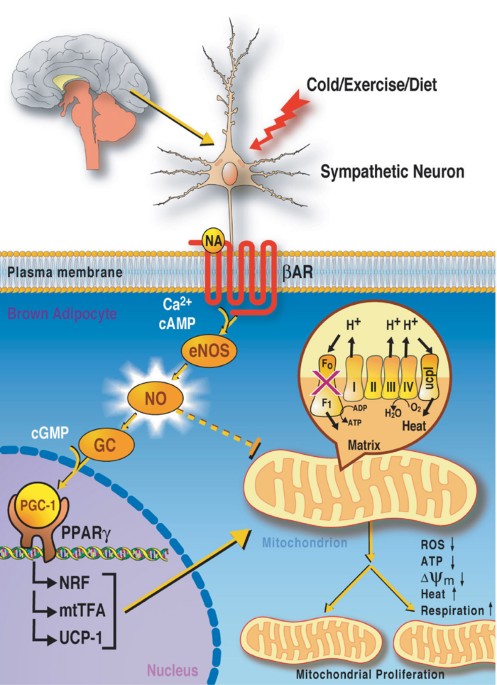
activation leads to Ca2+- and cAMP-mediated activation of endothelial NOS (eNOS) (Figure 1). In turn eNOS generates NO, which induces activation of the master regulator of mitochondrial
biosynthesis, PGC-1_α_ (peroxisome proliferator-activated receptor _γ_ coactivator-1_α_). PGC-1_α_ has been shown to be upregulated after cold exposure and changes in diet.1,2,3,4,5,6
Moreover, PGC-1_α_ transgenic mice exhibit increased numbers of mitochondria in heart and skeletal muscle.5 PGC-1_α_ is a key transcriptional cofactor of PPAR_γ_, a nuclear hormone receptor,
which increases the expression of nuclear respiratory factors (NRF) and the mitochondrial transcription factor A (mtTFA). This cascade of events increases the expression of nuclear and
mitochondrial genes encoding components of the respiratory chain complexes and regulators of mtDNA replication. Furthermore, antisense inhibition of PGC-1_α_ revealed that NO-induced
mitochondrial biogenesis is dependent on PGC-1_α_. Importantly, the effect of NO on adipose tissue is dependent on cyclic guanosine 3′, 5′-monophosphate (cGMP), thus making it less likely
that NO mediates mitochondrial biogenesis by directly affecting mitochondria. It is of note that NO has previously been shown to directly block mitochondrial respiration by competing for the
oxygen binding site of cytochrome c oxidase.12,13,14 Significantly, Nisoli _et al._7 now show that eNOS knockout mice exhibit a reduced number of BAT mitochondria, decreased UCP-1 and
PPAR-_γ_ expression, defective energy expenditure, increased body weight, insulin resistance, and hypertension.15,16,17 This is a phenotype characteristic of a metabolic syndrome and often
occurs during aging. _Extending lifespan: the insulin connection_. Is there a link between the NO-mediated pathway of mitochondrial proliferation proposed by Nisoli and colleagues and the
regulation of aging? In this regard, a number of observations might be of relevance. NO-mediated mitochondrial biogenesis promotes leanness due to adaptive thermogenesis and a reduction in
reactive oxygen species; both of these effects have previously been proposed to increase longevity (reviewed in Spiegelman and Flier18 and Guarente and Kenyon19). In contrast, as mentioned
above, eNOS knockout mice are insulin-resistant and manifest increased body weight.16 Thus, it is interesting to speculate that insulin signaling and NO-mediated mitochondrial biogenesis may
regulate both body weight and lifespan. Along these lines, levels of UCP proteins and mitochrondrial density are decreased during aging, while free radicals increase (reviewed in
Florez-Duquet and McDonald20). Paradoxically however, insulin receptor signaling has been proposed to accelerate aging (reviewed in Guarente and Kenyon19). Genetic studies on _Caenorabditis
elegans_ revealed that animals lacking DAF-2, a molecule with structural similarity to human insulin growth factor 1 (IGF-1) receptor, doubled their lifespan.21,22 In addition, Holzenberger
_et al._23 recently reported that mice heterozygous for IGF-1 receptor deficiency exhibit increased life expectancy. Insulin-related genes also have critical functions in growth and glucose
metabolism. Loss of insulin signaling results in dwarfism, diabetes, and obesity. Recently, however, Blüher _et al_ 24 reported that mice carrying a fat cell-specific insulin receptor
knockout (FIRKO) display enhanced longevity and reduced body weight, despite normal food intake. Caloric restriction has been proposed to counteract the aging process by decreasing
metabolism and free radical formation, but FIRKO mice manifest increased lifespan and are resistant to obesity, despite normal energy intake. Moreover, FIRKO mice display an increased rate
of metabolism. These findings imply that increased energy expenditure and metabolism may be more effective in the delay of aging process than a decreased food intake. _Bi-functionality of NO
in cell survival and cell death_. NO has the ability to either inhibit or promote cell death.25 How can the same molecule have apparently opposite functions? The molecular basis for this is
currently unclear. Several mechanisms have been proposed to account for the protective effects of NO. One such mechanism is the S-nitrosylation (or transfer of NO to a regulatory cysteine
thiol) of proteins involved in cell death, such as effector caspases, resulting in the inactivation of the caspase cascade.26,27 In addition, S-nitrosylation of NMDA receptors and other
proteins have been proposed to reduce cell death (reviewed in Lipton28). The observations of Nisoli _et al_ 7 suggest yet another mechanism whereby NO may promote survival, i.e. by
UCP-1-mediated uncoupling of mitochondria and reduction in free radical production. In contrast, excessive amounts of NO can combine with superoxide anions (O2−) to form toxic peroxynitrite
(ONOO−).29 While NO reversibly inhibits mitochondrial respiration,12,13,14 ONOO− irreversibly blocks many components of the mitochondrial respiratory chain. This process can trigger cell
death by apoptosis (if ATP levels are sufficient) or necrosis (if prolonged ATP deficits arise).30,31,32 Additionally, NO-mediated S-nitrosylation can activate metalloproteinases, thus
degrading the extracellualr matrix and promoting cell death by anoikis.33 Lastly, NO may trigger DNA damage and poly (ADP)-ribose synthetase (PARP)-mediated neuronal cell death.34
_Mitochondrial proliferation during cell death_. In contrast to NO-induced biogenesis of mitochondria in BAT, there is mounting evidence indicating that mitochondria also proliferate during
apoptotic cell death initiated by a wide variety of stimuli in a number of different cell types.35 Nevertheless, the role of mitochondrial proliferation during cell death remains
contentious. In fact, mitochondrial proliferation may constitute an early stress response to adapt to new energy demands and to repair injuries, thus promoting cell survival. Notably,
mitochondrial uncouplers, such as carbonylcyanide _p_-(trifluoromethyoxy) phenyl-hydrazone (FCCP), induce mitocho-ndrial proliferation, but do not induce cell death. It is currently unclear
whether UCP-1 and related factors play a role in the mitochondrial proliferation events observed during apoptosis. Along these lines, recent studies suggest that the brain mitochondrial
uncoupling protein-2 (UCP-2) exhibits protective effects against brain injuries.36 On the other hand, it has been suggested that mitochondrial proliferation, also referred to as
mitochondrial fission, might be an intricate part of the cell death program. Support for this notion is provided by the recent findings of Youle _et al._35 They report that overexpression of
a dominant-negative form of Drp-1, a highly conserved dynamin-related GTPase that is a component of the mitochondrial division/fission machinery, blocked apoptotic mitochondrial
proliferation, release of cytochrome _c_ from mitochondria, and cell death.35 In addition, Drp-1 and the proapoptotic Bax protein colocalized to scission sites on mitochondria, suggesting
that the mitochondrial fission machinery cooperates with the cell death machinery via Bcl-2 family members.37 Interestingly, NO can upregulate Bax and trigger its translocation to
mitochondria.38 Furthermore, NO-induced p38 MAP kinase activation has been shown to induce Bax translocation to mitochondria.39 Mitochondrial uncouplers promote Bax translocation to
mitochondria, suggesting that a decrease in mitochondrial membrane potential (Δ_ψ_m) may suffice to facilitate Bax membrane insertion.40 Thus, it is interesting to speculate that the dual
function of NO, in either promoting or preventing cell death, may have as part of its mechanistic basis the regulation of mitochondrial biogenesis. We propose that small amounts of NO may
stimulate mitochondrial proliferation, thereby conveying a survival signal. Conversely, excessive amounts of NO combined with free radicals may, in addition to generating mitochondria,
recruit the cell death machinery and thus commit cells to die. _Conclusions/future directions_. Mitochondrial biogenesis requires the choreographed expression of diverse transcriptional
activators, such as PGC-1, PPAR_γ_, NRF-1, and mtTFA. Nisoli _et al._7 have shown that NO acts as a key messenger to activate the mitochondrial biogenesis program (Figure 1). These findings
imply that NO regulates energy balance, may prevent obesity, and may play a role in determining lifespan. Thus, these results may provide new therapeutic opportunities to combat obesity and
associated diseases. In future experiments it will be interesting to test whether NO also regulates mitochondrial proliferation in other cell types and tissues. The question of lifespan in
eNOS knockout mice, eNOS transgenic mice, or PGC-1_α_ transgenic mice should also be explored. Lastly, it will be important to elucidate the precise mechanism by which NO/cGMP activates
PGC-1_α_ to trigger mitochondrial biogenesis. REFERENCES * Lowell BB, Spiegelman BM . (2000). _Nature_ 404: 652–660. Article CAS Google Scholar * Wu Z _et al_. (1999) _Cell_ 98: 115–124
Article CAS Google Scholar * Wu H _et al_. (2002) _Science_ 296: 349–352 Article CAS Google Scholar * Vibrasius JV, Scarpulla RC (1994) _Proc. Natl. Acad. Sci. USA_ 91: 1309–1313 *
Lehman JJ _et al_. (2000) _J. Clin. Invest_ 106: 847–856 * Scarpulla RC (2002) _Biochem. Biophys. Acta_ 1576: 1–14 * Nisoli E _et al_. (2003) _Science_ 299: 896–899 Article CAS Google
Scholar * Nisoli E _et al_. (1998) _Brit. J. Pharm_ 125: 888–894 * Boss O _et al_. (1999) _Biochem. Biophys. Res. Commun_ 261: 870–876 * Bredt DS, Snyder SH (1994) _Ann. Rev. Biochem_ 63:
175–195 * Gow AJ, Stamler JS (1998) _Nature_ 391: 169–173 Article CAS Google Scholar * Cleeter MWJ _et al_. (1994) _FEBS Lett_ 345: 50–54 * Schweizer M, Richter C (1994) _Biochem.
Biophys. Res. Commun_ 204: 169–175 * Brown GC, Cooper CE (1994) _FEBS Lett_ 356: 295–298 * Momken I _et al_. (2002) _Biochem. J_ 368: 341–347 * Shankar RR _et al_. (2000) _Diabetes_ 49: 1–4
* Huang PL _et al_. (1995) _Nature_ 377: 239–242 Article CAS Google Scholar * Spiegelman BM, Flier JS (2001) _Cell_ 104: 531–543 Article CAS Google Scholar * Guarente L, Kenyon C
(2000) _Nature_ 403: 255–262 Article CAS Google Scholar * Florez-Duquet M, McDonald RB (1998) _Physiol. Rev_ 78: 339–358 * Kenyon C _et al_. (1993) _Nature_ 366: 461–464 Article CAS
Google Scholar * Kimura KD _et al_. (1997) _Science_ 277: 949–946 * Holzenberger M _et al_. (2003) _Nature_ 421: 182–186 * Blüher M _et al_. (2003) _Science_ 299: 572–574 Article Google
Scholar * Lipton SA _et al_. (1993) _Nature_ 364: 626–632 Article CAS Google Scholar * Melino G _et al_. (1997) _Nature_ 388: 432–433 Article CAS Google Scholar * Tenneti L _et al_.
(1997) _Neurosci. Lett_ 236: 139–142 * Lipton SA (1999) _Cell Death Differ_ 6: 943–951 Article CAS Google Scholar * Stamler JS _et al_. (1992) _Science_ 258: 1898–1902 Article CAS
Google Scholar * Cassina A, Radi R (1996) _Arch. Biophys. Biochem_ 328: 309–316 * Clementi E _et al_. (1998) _Proc. Natl. Acad. Sci. USA_ 95: 7631–7636 * Bonfoco E _et al_. (1995) _Proc.
Natl. Acad. Sci. USA_ 92: 7162–7166 * Gu Z _et al_. (2002) _Science_ 297: 1186–1190 Article CAS Google Scholar * Zhang J _et al_. (1994) _Science_ 263: 687–689 Article CAS Google
Scholar * Frank S _et al_. (2001) _Dev. Cell_ 1: 515–525 * Bechman I _et al_. (2002) _Biochem. Pharmacol_ 64: 363–367 * Karbowski M _et al_. (2002) _J. Cell Biol_ 159: 931–938 * Tamatani M
_et al_. (1998) _J. Neurochem_ 71: 1588–1596 * Ghatan S _et al_. (2000) _J. Cell Biol_ 150: 335–347 * Smaili SS _et al_. (2001) _Cell Death Differ_ 8: 909–920 Article CAS Google Scholar
Download references ACKNOWLEDGEMENTS We thank K. Gramatikoff for help with graphics. This work was supported by NIH Grants R01 NS 44314 to E.B.-W. and P01 HD29587, R01 EY05477, R01 EY09024,
and R01 NS41207 to S.A. L. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Del E. Webb Center for Neuroscience and Aging, The Burnham Institute, 10901 North Torrey Pines Rd., La Jolla, 92037,
CA, USA E Bossy-Wetzel & S A Lipton Authors * E Bossy-Wetzel View author publications You can also search for this author inPubMed Google Scholar * S A Lipton View author publications
You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to E Bossy-Wetzel. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE
THIS ARTICLE Bossy-Wetzel, E., Lipton, S. Nitric oxide signaling regulates mitochondrial number and function. _Cell Death Differ_ 10, 757–760 (2003). https://doi.org/10.1038/sj.cdd.4401244
Download citation * Published: 18 June 2003 * Issue Date: 01 July 2003 * DOI: https://doi.org/10.1038/sj.cdd.4401244 SHARE THIS ARTICLE Anyone you share the following link with will be able
to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing
initiative
Trending News
Error 404 | COPELo sentimos, no encontramos la página o enlace al que intenta acceder.INSTITUCIONALCOPE revalida su compromiso de servir...
Four quick language tips to help you in everyday frenchTHESE HANDY REMINDERS WILL MAKE NAVIGATING CONVERSATIONS EASIER Learning French can be both a rewarding and a tiring exp...
How to add liquidity to viper swap liquidity pools: step by step guideYield farming allows you to make more money with your crypto assets by becoming a Liquidity Provider on Viper Swap. When...
Only in America | The WeekPeople for the Ethical Treatment of Animals has asked that a man awaiting trial for cannibalism be forced to become vege...
Disruption by carcinogens of the hormone dependent association of membranes with polysomesABSTRACT It can be shown by enzyme assay that the effect of a variety of carcinogens is the degranulation of the endopla...
Latests News
Nitric oxide signaling regulates mitochondrial number and functionObesity affects many millions of children and adults worldwide and poses a major health problem. Weight gain results whe...
Bhilwara election result 2024 live updates: bjp's damodar agarwal has won this lok sabha seatBHILWARA LOK SABHA ELECTION RESULT 2024 LIVE UPDATES: With the counting of votes for the 2024 Lok Sabha elections underw...
MarketWatch Jack Denton is a reporter with Barron's Group in London. He writes about business and finance in Europe for Barron's and MarketWatch, withMarketWatch Jack Denton is a reporter with Barron's Group in London. He writes about business and finance in Europe...
Andrew C. PorterAndrew C. Porter will become the dean of the graduate school of education at the University of Pennsylvania, in Philadel...
Hispanic transgender rights activist sylvia rivera — aarp vivaEn español | A 17-year-old Puerto Rican-Venezuelan transgendered woman named Sylvia Rivera helped kick off the Stonewall...