Origins of hyperphenylalaninemia in israel
Origins of hyperphenylalaninemia in israel"
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Mutations and polymorphisms at the phenylalanine hydroxylase (PAH) gene were used to study the genetic diversity of the Jewish and Palestinian Arab populations in Israel. PAH
mutations are responsible for a large variety of hyperphenylalaninemias (HPAs), ranging from the autosomal recessive disease phenylketonuria to various degrees of nonclinical HPA.
Seventy-two Jewish and 36 Palestinian Arab families with various HPAs, containing 115 affected genotypes, were studied by haplotype analysis, screening for previously known PAH lesions and a
search for novel mutations. Forty-one PAH haplotypes were observed in this sample. Four mutations previously identified in Europe (IVS10nt546, R261Q, R408W and R158Q) were found, and were
associated with the same haplotypes as in Europe, indicating possible gene flow from European populations into the Jewish and Palestinian gene pools. Of particular interest is a PAH allele
with the IVS10nt546 mutation and haplotype 6, that might have originated in Italy more than 3,000 years ago and spread during the expansion of the Roman Empire. These results, together with
previous identification of three PAH mutations unique to Palestinian Arabs [IVSnt2, Edel(197–205) and R270S], indicate that the relatively high genetic diversity of the Jewish and
Palestinian populations reflects, in addition to genetic events unique to these communities, some gene flow from neighboring and conquering populations. You have full access to this article
via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS ALLELE FREQUENCY OF VARIANTS REPORTED TO CAUSE ADENINE PHOSPHORIBOSYLTRANSFERASE DEFICIENCY Article Open access 11
March 2021 TRACING THE MUTATED _HTT_ AND HAPLOTYPE OF THE AFRICAN ANCESTOR WHO SPREAD HUNTINGTON DISEASE INTO THE MIDDLE EAST Article 14 July 2020 CLINICAL AND MOLECULAR CHARACTERIZATION OF
PRIMARY HYPEROXALURIA IN EGYPT Article Open access 23 September 2022 INTRODUCTION Characterization of mutant genes associated with hereditary diseases is aimed primarily at understanding the
molecular basis of these disorders. However, identification of sequence variations associated with normal and defective alleles can also contribute to our understanding of the mechanisms
underlying the dynamics of gene flow among human populations. Alleles marked by mutations or polymorphic haplotypes or a combination of both may serve as useful markers to study the
interrelationships between various ethnic groups and the origins of their genetic diversity. The gene encoding the hepatic enzyme phenylalanine hydroxylase (PAH) has recently been a focus of
such investigations. PAH catalyzes the irreversible hydroxylation of phenylalanine to tyrosine, thereby opening the catabolic pathway of this essential amino acid. Mutations in this gene
cause a wide spectrum of disorders collectively called hyperphenylalaninemias (HPAs) [1]- Extreme HPA is expressed as the autosomal recessive disease phenylketonuria (PKU), which causes
severe mental retardation. Early diagnosis of PKU and subsequent treatment with a low-phenylalanine diet can considerably reduce the brain damage, making newborn screening for HPA a common
practice in many countries. Milder HPAs have no clinical consequences and are usually identified by biochemical tests. Extensive studies in European, Mediterranean and Asian communities
recently disclosed a wide variety of PAH mutations underlying these conditions [see ref. 2, 3 for reviews]. Mutations which affect PAH activity severely result in PKU in homozygotes or
compound heterozygotes, while non-PKU HPA is usually caused by compound heterozygosity for a mutation of the severe type and a mutation with a milder effect on the protein [4–7]. PAH
mutations show strong linkage disequilibrium with specific polymorphic haplotypes at the PAH locus [1–3]. Thus, mutant PAH genes can be ‘tagged’ with specific mutation-haplotype
combinations, which enable one to follow their spread in different populations with great specificity [8]. More than 2 million Israeli newborns have been screened for HPA since 1960. PAH
deficiencies were identified in some 450 infants, 30% of them showing PKU and the rest non-PKU HPA [9–11; unpubl. data]. Both types of HPA were identified among Jews and Palestinian Arabs.
These two populations differ from the European and Asian communities studied thus far, and from each other. The Jewish population is composed mostly of immigrants from a variety of
countries, and their descendents, while the Palestinian Arab population reflects the migrations and foreign conquests that have swept this country since the time of the Roman Empire. Both
ethnic groups are therefore expected to show considerable genetic diversity for their small size. We are tracing the origins of this diversity, using as a probe the mutations and
polymorphisms of the PAH gene. SUBJECTS AND METHODS SAMPLE POPULATION The population of individuals affected with HPAs in Israel, and their ascertainment, have been described previously [4,
9–11]. The great majority of HPA cases in the Jewish population was identified among Sephardic and Oriental Jews, rather than in Ashkenazi communities [9, 10]. Over a period of 5 years, we
collected peripheral blood samples from 72 Jewish and 36 Palestinian families with HPAs. Since our primary interest was directed towards PKU, in most of these families (a total of 88) the
probands had PKU. In 13 families non-PKU HPA was identified, and in 7 families both conditions were segregating [see ref. 4 for a detailed description of these families]. Altogether, 115
affected genotypes were studied (each of the 7 families with both PKU and non-PKU HPA contributed 2 affected genotypes). Among the Jewish families, 65 were of Sephardic or Oriental origin, 5
were Ashkenazi and 2 represented ‘mixed marriages’. The parents were consanguineous in 34 of the 36 Palestinian families and in 6 of the 72 Jewish families. MOLECULAR METHODS Extraction of
genomic DNA from peripheral blood samples, haplotype analysis, identification of previously known mutations and the search for new mutations have been previously described in detail [4,
12–16]. RESULTS POLYMORPHIC HAPLOTYPES AT THE PAH LOCUS Eight RFLPs conveniently detected by the full cDNA of PAH [17, 18] were used to construct polymorphic PAH haplotypes in all the
families studied. Forty-one haplotypes were observed in the sample population. Their distribution among normal and mutant PAH genes is shown in table 1. Complete haplotype analysis was not
possible in some families, since not all parents were available. In other families, heterozygosity of all family members for one or two RFLPs and unavailability of additional family members
precluded haplotype construction. Hence, the final number of haplotyped chromosomes is slightly smaller than expected on the basis of the sample size. Except for haplotype 6del which is
unique to Yemenite Jewish PKU patients [19], all of the haplotypes identified in this study have also been observed in European and Mediterranean populations [2, 3, 8, 18, 20]. Although a
considerable variety of haplotypes was found among Jews and Palestinian Arabs, in both groups most of the haplotypes occurred on a single or small number of chromosomes, with three or four
haplotypes predominant. Haplotypes 1, 4 and 6 were abundant among both Jews and Palestinians; haplotype 28 was also frequent among Jews, as was the ‘Yemenite’ PKU haplotype, 6del. For
haplotypes other than 6del, χ2 analysis indicated significant difference between frequencies among Jews and Palestinian Arabs only for mutant haplotype 1. Most haplotypes were shared by
wild-type and mutant alleles, and only two haplotypes in Jews and two in Palestinian Arabs were unique to mutant alleles (table 1). Haplotype 6 was found almost exclusively on mutant
chromosomes in both groups. The major haplotypes on mutant alleles among Jews were 6del, 1, 4, 6 and 7 (table 1). While more than half of the mutations associated with haplotypes 4 and 6
originated in North African communities (table 2), mutant haplotype 1 was found mainly in Jewish patients from Middle Eastern countries. POINT MUTATIONS IN THE PAH GENE Tables 3 and 4
summarize PAH molecular lesions found to date in the Israeli population, their haplotype association and ethnic origins. The entire patient population — excluding the Yemenite Jewish PKU
patients who are all homozygous for a deletion of PAH exon 3 [19] — was screened for the presence of eleven point mutations previously identified in European and Mediterranean populations:
R158Q, R252W, R261Q, G272X, S273F, E280K, P281L, L31 1P, IVS10nt546, R408W and Y414C [2, 3]. Four of these mutations were identified on 50 of the 168 mutant alleles tested; 35 of the 50
alleles contained the splicing defect IVS10nt546 (table 3). Notably, the haplotype association of these mutations (table 4) was similar in the Israeli and European populations, with the
exception of two alleles carrying the IVS10nt546 mutation on the background of haplotype 5, found in a Palestinian Arab patient. This association suggests that several mutant PAH alleles
among Jews and Palestinians share common origins with similar alleles in these countries. A search for novel point mutations in the sample population has yielded thus far four mutations:
E6del(197–205) [13] IVS2ntl [14], R270S [16] and S349P [12]. The first three mutations are unique to Palestinian Arabs and were not reported in any other population, while S349P was
discovered independently in French-Canadian and European patients [21, 22]. Haplotype association of these mutations is shown in table 4. Two mutations were identified in 20 of 28 mutant PAH
genes among Moroccan Jewish patients: IVS10nt546, which was associated mostly with haplotype 6, and on one chromosome with haplotype 36; and S349P, all associated with haplotype 4 (table
4). The origin of these chromosomes in this community was studied by screening 17 Moslem Moroccan patients for the same mutations. The IVS10nt546 mutation was found in one patient, in
association with haplotypes 6 and 36, and S349P was identified in another one, with haplotype 4. DISCUSSION The variety of polymorphisms and mutations at the PAH locus makes it a useful tool
in molecular population genetics. Due to the strong linkage disequilibrium between PAH mutations and specific haplotypes, it is highly probable that alleles carrying the same
mutation-haplotype combination are identical in descent. This notion allows one to trace the origin of such alleles back in time and geography, and to uncover mechanisms underlying the
genetic diversity or genetic similarity of different communities. Typical examples are the identification of a single PAH deletion responsible for all PKU cases among Yemenite Jews, which
was traced back to specific families several hundred years ago [19], and the recent localization of the origins of several PAH mutations to specific European ethnic subgroups [8]. The two
populations investigated in this study are very different from each other: The Jewish population of Israel is composed mostly of immigrants who have come from numerous countries since the
beginning of the 20th century, and their descendents. These people have ancient roots in this region that date back to the Jewish Kingdom of the first and second millenia BC. The spread of
the Jewish people around the world following the destruction of the Kingdom and deportation by the Romans in the first century AD should have resulted in increased genetic diversity despite
their relative genetic seclusion in the diaspora. The Palestinian Arab population originated in Arab tribes that migrated to Palestine from the Arabian Peninsula at the time of the
Roman-Byzantine Empire, and expanded during the Arab-Islamic conquest of the country. The Palestinian Arabs have been residing in this country since that time, living under different rulers
and assimilating various waves of immigrants, including the crusaders and the Turks. Haplotype analysis in this study was based on eight RFLPs [17] which served to establish a commonly used
haplotype nomenclature [18]. Additional polymorphisms are being discovered at the PAH locus [23–25], and their incorporation into the existing haplotype system should refine this
nomenclature [20]. It should be noted that the subjects studied do not constitute random samples of their communities, since only families with HPAs were selected for analysis. A relatively
large number of different haplotypes was found in the Jewish and Palestinian Arab populations in relation to their size, reflecting considerable genetic variability. But, a small number of
haplotypes account for the majority of PAH alleles identified. The geographic origins of the mutant PAH haplotypes in the Israeli Jewish population summarized in table 2 show that the
majority of HPA cases among Jews in Israel are in families of north African, Additional Eastern and Asian origin. It is noteworthy that in some cases the same haplotype appears on mutant
alleles from countries in different parts of the world, e.g., haplotypes 1, 4, 6 and 7. However, haplotypes alone, particularly the more abundant ones, give only a weak indication of genetic
similarity and can be used to pinpoint genetic relationships between different communities only when the associated mutations are identified [26]. Haplotype 1, for example, was found to
harbor at least 14 different mutations in Europe [2]. It is noteworthy in this regard that mutations common to the Israeli and European populations are associated in most cases with the same
haplotypes (table 3), indicating possible common origins. Three of these mutations — R158Q, R261Q and R408W — are associated with the dinucleotide CpG, a known hot spot for mutations [27],
and therefore recurrence of these mutations cannot be ruled out [28, 29]. However, it seems to us that the similar haplotype association favors the possibility of common origin, particularly
in the case of the R408W/haplotype 2 allele which is rare in the Israeli population (table 4). It is not surprising to see this allele in Jews from eastern Europe (table 4), since it has a
Balto-Slavic origin and accounts for a high proportion of PKU alleles in eastern Europe and Germany [2, 8, 30–35]. It is interesting to note the appearance of this allele and the
R158Q/haplotype 4 combination in Palestinians (table 4). Gene flow from Europe into the Palestinian gene pool could have occurred at various periods in history: during the waves of migration
from western and central Europe (including the Balkan region) in the 1st century BC, during the Roman conquest of the region up to the Arab-Islamic conquest in the 7th century AD, and
during the Crusades from 1099 AD through the 13th century. Additionally, Christian pilgrimages to the Holy Land were continuous throughout the last 20 centuries. Most prominent on the list
of mutant PAH alleles common to Israeli and other populations is IVS10nt546/haplotype 6 (tables 3, 4). It shows a unique and widespread geographic distribution, with particular prevalence in
eastern Europe and Turkey, and has also been reported in Italy, Spain and western Europe [31, 36–41]. Romano et al. [41] detected this allele in the eastern part of Sicily and concluded
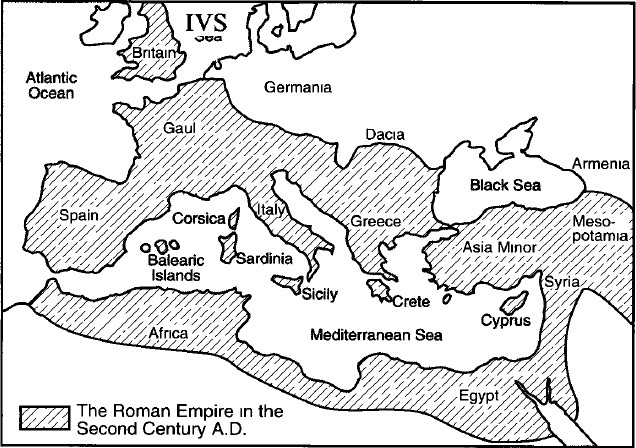
that the origin was probably Italic, before the 10th century BC. A compilation of the geographic locations of the IVS10nt546/haplotype 6 allele based on this and previous studies leads us to
suggest that this allele may indeed have originated in Italy before the first millenium BC, and spread through the Mediterranean littoral countries and eastern Europe during the expansion
of the Roman Empire in the first century BC. The regions included in that Empire at its height in the 2nd century AD (fig. 1) roughly correlate with the current prevalence map of this
allele. The appearance of the mutation in Palestinian Arabs may reflect the Roman and subsequent Byzantine centuries-long rule in Palestine. The local Christian population assumed the
Islamic religion during the Arab conquest in the 7th century, and was totally assimilated into the Arab population. The predominance of the IVS10nt546/haplotype 6 chromosome among Moroccan
Jews is intriguing. The frequency of this chromosome in our Moslem sample population is low (1/34) but, interestingly, the only other IVS10nt546 chromosome in the Moslem sample carries
haplotype 36 found with this mutation in one Jewish patient. Another PAH allele found in one Moslem patient and among the Jewish patients in Morocco is S349P/haplotype 4. Jewish-Moslem
intermarriages were extremely rare in this country, being strictly forbidden by the Jewish religion. A possible source of the mutation in the Jewish gene pool might be the conversion to
Judaism of local populations, including the Berbers, that took place during the last centuries of Roman and Byzantine rule in the area, before the Arab-Islamic conquest at the end of the 7th
century. The few IVS10nt546/haplotype 6 alleles found in Jews from Asian countries (table 4) can be attributed to occasional migrations eastward of Jews fleeing religious persecution under
Roman/Byzantine rule. A similar conclusion may be drawn for the R261Q/haplotype 1 allele which, however, does not penetrate the Moroccan-Jewish gene pool. A clear example of a recurrent
mutation in our population is S349P. This mutation was identified independently in an Australian PKU patient for whom the haplotype association was not reported [21], and in French-Canadian
patients in association with haplotype 1 [22]. The same mutation was discovered independently in our laboratory in Jewish patients of north African extraction, on the background of haplotype
4 [12]. The recurrence of this mutation is particularly interesting because it does not involve a CpG dinucleotide. A search for novel mutations among Palestinian PKU patients has led to
the identification of 3 ‘private’ mutations not shared by other populations (table 3). It is still possible that these mutations did not occur within the Palestinian community, and might be
common to other Arab communities in the region — a question that can be resolved by screening PKU patients in neighboring countries. The usefulness of PAH genotypes as population genetic
markers should increase considerably once the entire spectrum of HPA mutations is revealed and haplotype nomenclature further refined by additional polymorphisms. We expect these sequence
variations to disclose additional, unknown origins accounting for the genetic diversity of the peoples of our region. REFERENCES * Scriver CR, Kaufman S, Woo SLC: The hyperphenylalaninemias;
in Scriver CR, Beadet AL, Sly WS, Valle D (eds): The Metabolic Basis of Inherited Disease, ed 6. New York, McGraw-Hill, 1989, pp 495–546. Google Scholar * Eisensmith RC, Woo SLC: Molecular
basis of phenylketonuria and related hyperphenylalaninemias: Mutations and polymorphisms in the human phenylalanine hydroxylase gene. Hum Mutat 1992;1:13–23 Article CAS PubMed Google
Scholar * Scriver CR, John SMW, Rozen R, Eisensmith R, Woo SLC: Association between populations, phenylketonuria mutations and RFLP haplotypes at the phenylalanine hydroxylase locus: An
overview. Dev Brain Dysfunct 1993;6:11–25 Google Scholar * Avigad S, Kleiman S, Weinstein M, Cohen BE, Schwartz G, Woo SLC, Shiloh Y: Compound heterozygosity in non-PKU
hyperphenylalaninemia: The contribution of mutations for classical PKU. Am J Hum Genet 1991;48:393–399 Google Scholar * Okano Y, Eisensmith RC, Guttler F, Lichter-Konecki U, Konecki DS,
Trefz FK, Dasovich M, Wang T, Henriksen K Lou H, Woo SLC: Molecular basis of phenotypic heterogeneity in PKU. N Engl J Med 1991;324:1232–1238 Article CAS PubMed Google Scholar *
Economou-Petersen E, Henriksen KF, Guldberg P, Guttler F: Molecular basis for nonphenylketonuria hyperphenylalaninemia. Genomics 1992;14:1–5 Article CAS PubMed Google Scholar * Svensson
E, Eisensmith RC, Dworniczak B, von Dobeln U, Hagenfeldt L, Horst J, Woo SLC: Two missense mutations causing mild hyperphenylalaninemia associated with DNA haplotype 12. Hum Mutat
1992;1:129–137 Article CAS PubMed Google Scholar * Eisensmith RC, Okano Y, Dasovich M, Wang T, Guttler F, Lou H, Guldberg P, Lichter-Konecki U, Konecki DS, Svensson E, Hagenfeldt L, Rey
F, Munnich A, Lyonnet S, Cockburn F, Conor JM, Pembrey ME, Smith I, Gitzelmann R, Steinmann B, Apold J, Eiken HG, Giovannini M, Riva E, Longhi C, Romano V, Gerone R, Naughten ER, Mullins C,
Cahalane S, Ozalp I, Fekete G, Schuler D, Berencsi GY, Nasz I, Brdicka R, Kamaryt J, Pijackova A, Cabalska B, Boszkowa K, Schwartz E, Kalinin VN, Jin L, Chakraborty R, Woo SLC: Multiple
origins for phenylketonuria in Europe. Am J Hum Genet 1993;51:1355–1365 Google Scholar * Cohen BE, Bodoyni E, Szeinberg A: Phenylketonuria in Jews. Lancet 1961;i:344–345 Article Google
Scholar * Cohen BE, Szeinberg A, Levine T, Peled I, Pollack S, Crispin M, Normand M: Phenylketonuria in Israel. Monogr Hum Genet 1978;9:95–101 Article CAS PubMed Google Scholar * Shiloh
Y, Avigad S, Kleiman S, Weinstein M, Schwartz G, Woo SLC, Cohen BE: Molecular analysis of hyperphenylalaninemia in Israel: A study of Jewish genetic diversity; in Bonne-Tamir B, Adam A
(eds): Genetic Diversity among Jews: Diseases and Markers at the DNA Level. Oxford, Oxford University Press, 1992, pp 237–247. Google Scholar * Weinstein M, Eisensmith RC, Abadie V, Avigad
S, Lyonnet S, Schwartz G, Munnich A, Woo SLC, Shiloh Y: A missense mutation, S349P, completely inactivates phenylalanine hydroxylase in North African Jews with phenylketonuria. Hum Genet
1993;90:645–649 Article CAS PubMed Google Scholar * Kleiman S, Schwartz G, Akawi Y, Woo SLC, Shiloh Y: A 22-bp deletion at the phenylalanine hydroxylase gene causing phenylketonuria in
an Arab family. Hum Mutat 1992;1:344–346 Article CAS PubMed Google Scholar * Kleiman S, Bernstein J, Schwartz G, Woo SLC, Shiloh Y: A defective splice site at the phenylalanine
hydroxylase gene in phenylketonuria and benign hyperphenylalaninemia among Palestinian Arabs. Hum Mutat 1992;1:340–343 Article CAS PubMed Google Scholar * Kleiman S, Bernstein J,
Schwartz G, Brand N, Elitzur A, Woo SLC, Shiloh Y: Phenylketonuria: Variable phenotypic outcomes of the E261Q mutation, and maternal PKU in the offspring of a healthy homozygote. J Med Genet
1993;30:284–288 Article CAS PubMed PubMed Central Google Scholar * Kleiman S, Li J, Schwartz G, Eisensmith RC, Woo SLC, Shiloh Y: Inactivation of phenylalanine hydroxylase by a
missense mutation, R270S, in a Palestinian kinship with phenylketonuria. Hum Mol Genet 1993;2:605–606 Article CAS PubMed Google Scholar * Lidsky AS, Ledley FD, DiLella AG, Kwok SCM,
Daiger SP, Robson KJH, Woo SLC: Extensive restriction site polymorphism at the human phenylalanine hydroxylase locus and application in prenatal diagnosis of phenylketonuria. Am J Hum Genet
1985;37:619–634 CAS PubMed PubMed Central Google Scholar * Woo SLC: Collation of RFLP haplotypes at the human phenylalanine hydroxylase (PAH) locus. Am J Hum Genet 1988;43:781–783 CAS
PubMed PubMed Central Google Scholar * Avigad S, Cohen BE, Bauer S, Schwartz G, Frydman M, Woo SLC, Niny Y, Shiloh Y: A single origin of phenylketonuria in Yemenite Jews. Nature
1990;344:168–170 Article CAS PubMed Google Scholar * Eisensmith RC, Woo SLC: Updated listing of haplotypes at the human phenylalanine hydroxylase (PAH) locus. Am J Hum Genet
1992;51:1445–1448 CAS PubMed PubMed Central Google Scholar * Forrest SM, Dahl HH, Howells DW, Dianzani I, Cotton RGH: Mutation detection in phenylketonuria by using chemical cleavage of
mismatch: Importance of using probes from both normal and patient samples. Am J Hum Genet 1991;49:175–183 CAS PubMed PubMed Central Google Scholar * John SWM, Rozen R, Laframboise R,
Laberge C, Scriver CR: Five mutations at the PAH locus account for almost 90% of PKU mutations in French-Canadians from Eastern Quebec. Hum Mutat 1992;1:72–74 Article CAS PubMed Google
Scholar * Kalaydjieva L, Dworniczak B, Aulehla-Scholz C, Devoto M, Romeo G, Stuhrmann M, Kuchinskas V, Yurgelyavicius V, Horst J: Silent mutations in the phenylalanine hydroxylase gene as
an aid to the diagnosis of phenylketonuria. J Med Genet 1991;28:686–690 Article CAS PubMed PubMed Central Google Scholar * Golstov AA, Eisensmith RC, Konecki DS, Lichter-Konecki U, Woo
SLC: Association between mutations and a VNTR in the human phenylalanine hydroxylase gene. Am J Hum Genet 1992,51:627–636. Google Scholar * Golstov AA, Eisensmith RC, Naughton E, Jin L,
Chakraborty R, Woo SLC: A single polymorphic STR system in the human phenylalanine hydroxylase gene permits rapid prenatal diagnosis and carrier screening for phenylketonuria. Hum Mol Genet
1993;2:577–581 Article Google Scholar * Treacy E, Byck S, Clow C, Scriver CR: ‘Celtic’ phenylketonuria chromosomes found? Evidence in two regions of Quebec province. Eur J Hum Genet
1993;1:220–228 Article CAS PubMed Google Scholar * Abadie V, Lyonnet S, Maurin N, Berthelon M, Caillaud C, Giraud F, Mattei JF, Rey J, Rey F, Munnich A: CpG dinucleotides are mutation
hot spots in phenylketonuria. Genomics 1989;5:936–939 Article CAS PubMed Google Scholar * John SWM, Rozen R, Scriver CR, Laframbroise R, Laberge C: Recurrent mutation, gene conversion,
or recombination at the human phenylalanine hydroxylase locus: Evidence in French-Canadians and a catalog of mutations. Am J Hum Genet 1990;46:970–974 CAS PubMed PubMed Central Google
Scholar * Okano Y, Wang T, Eisensmith RC, Guttler F, Woo SLC: Recurrent mutation in the human phenylalanine hydroxylase gene. Am J Hum Genet 1990;46:919–924 CAS PubMed PubMed Central
Google Scholar * Kalaydjieva L, Dworniczak B, Aulehla-Scholz C, Kremensky I, Bronzova J, Eigel A, Horst J: Classical phenylketonuria in Bulgaria: RFLP haplotypes and frequency of the major
mutations. J Med Genet 1990;27:742–745 Article CAS PubMed PubMed Central Google Scholar * Kalaydjieva L, Dworniczak B, Kremensky I, Radeva B, Horst J: Population genetics of
phenylketonuria in Bulgaria. Dev Brain Dysfunct 1993;6:39–45 Google Scholar * Horst J, Eigel A, Kalaydjieva L, Dworniczak B: Phenylketonuria in Germany — molecular heterogeneity and
diagnostic implications. Dev Brain Dysfunct 1993;6:32–38 Google Scholar * Jaruzelska J, Henriksen KF, Guttler F, Riess O, Borski K, Blin N, Slomski R: The codon 408 mutation associated with
haplotype 2 is predominant in Polish families with phenylketonuria. Hum Genet 1991;86:247–250 Article CAS PubMed Google Scholar * Zygulska M, Eigel A, Dworniczak B, Sutkowska A,
Pietrzyk J, Horst J: Phenylketonuria in Poland: 66% of PKU alleles are caused by three mutations. Hum Genet 1991;88:91–94 Article CAS PubMed Google Scholar * Zygulska M, Eigel A,
Dworniczak B, Pietrzyk JJ, Horst J: Molecular analysis of phenylketonuria in the population of southern Poland. Dev Brain Dysfunct 1993;6:127–133 Google Scholar * Kalaydjieva L, Dworniczak
B, Aulehla-Scholz C, Devoto M, Romeo G, Stuhrmann M, Horst J: Phenylketonuria mutation in southern Europeans. Lancet 1991;i:865. Article Google Scholar * Dworniczak B, Aulehla-Scholz C,
Kalaydjieva L, Bartholome K, Grudda K, Horst J: Aberrant splicing of phenylalanine hydroxylase mRNA: The major cause for phenylketonuria in parts of southern Europe. Genomics 1991;11:242–246
Article CAS PubMed Google Scholar * Dasovich M, Konecki D, Lichter-Konecki U, Eisensmith RC, Guttler F, Naughton E, Mullins C, Giovannini M, Woo SLC: Molecular characterization of PKU
allele prevalent in southern Europe and Ireland. Somat Cell Mol Genet 1991;17:303–309 Article CAS PubMed Google Scholar * Perez B, Desviat LR, Die M, Ugarte M: Mutation analysis of
phenylketonuria in Spain: Prevalence of two Mediterranean mutations. Hum Genet 1992;89:341–342 Article CAS PubMed Google Scholar * Tyfield LA, Osborn MJ, King SK, Jones MM, Holton JB:
Molecular basis of phenylketonuria in an English population. Dev Brain Dysfunct 1993;6:60–67 Google Scholar * Romano V, Bosco P, Chiavetta V, Fasulo G, Pitronaci L, Mollica F, Meli C,
Giovannini M, Riva E, Giuffre B, Eisensmith R, Woo SLC, Romano C, Ponzone A, Dianzani I, Camaschella C, Di Pietro C, Ceratto N: Geographical distribution of phenylalanine hydroxylase alleles
in Sicily. Dev Brain Dysfunct 1993;6:83–91 Google Scholar * Okano Y, Wang T, Eisensmith RC, Longhi R, Riva E, Giovannini M, Cerone R, Romano C, Woo SLC: Phenylketonuria missense mutations
in the Mediterranean. Genomics 1991;9:96–103 Article CAS PubMed Google Scholar * Dianzani I, Forrest SM, Camaschella C, Saglio G, Ponzone A, Cotton GH: Screening for mutations in the
phenylalanine hydroxylase gene from patients with phenylketonuria by using the chemical cleavage method: A new splice mutation. Am J Hum Genet 1991;48:631–635 CAS PubMed PubMed Central
Google Scholar * Caillaud C, Vilarinho L, Vilarinho A, Rey F, Bertheion M, Santos R, Lyonnet S, Briard ML, Osorio RV, Rey J, Munnich A: Linkage disequilibrium between phenylketonuria and
RFLP haplotype 1 at the phenylalanine hydroxylase locus in Portugal. Hum Genet 1990;89:69–72 Article Google Scholar * Bertheion M, Caillaud C, Rey F, Labrune P, Melle D, Feingold J, Frezal
J, Briard ML, Farriaux JP, Guibaud P, Journel H, Maurin N, Le Marrec B, Nivelon JL, Plauchu H, Saudubray JM, Tron P, Rey J, Munnich A, Lyonnet S: Spectrum of phenylketonuria mutations in
Western Europe and North Africa, and their relation to polymorphic DNA haplotypes at the phenylalanine hydroxylase locus. Hum Genet 1991;86:355–358. Article Google Scholar * Ozguc M, Ozalp
I, Coskun T, Yilmaz E, Erdem H, Ayter S: Molecular analysis in Turkish phenylketonuria patients. J Med Genet 1993;30:129–130 Article CAS PubMed PubMed Central Google Scholar * Abadie
V, Lyonnet S, Melle D, Berthelon M, Caillaud C, Labrune P, Rey F, Rey J, Munnich A: Molecular basis of phenylketonuria in France. Dev Brain Dysfunct 1993;6:120–126 Google Scholar * Vereist
P, Francois B, Cassiman JJ, Raus J: Heterogeneity of phenylketonuria in Belgium. Dev Brain Dysfunct 1993;6:97–109 Google Scholar * DiLella AG, Marvit J, Lidsky AS, Guttler F, Woo SLC: Tight
linkage between a splicing mutation and a specific DNA haplotype in phenylketonuria. Nature 1986;322:799–803 Article CAS PubMed Google Scholar * Lyonnet S, Caillaud C, Rey F, Berthelon
M, Frezal J, Rey J, Munnich A: Molecular genetics of phenylketonuria in Mediterranean countries: A mutation associated with partial phenylalanine hydroxylase deficiency. Am J Hum Genet
1978;44:511–517 Google Scholar * Dworniczak B, Aulehla-Scholz C, Horst J: Phenylketonuria: Detection of a frequent haplotype 4 allele mutation. Hum Genet 1989;84:95–96 Article CAS PubMed
Google Scholar * Okano Y, Eisensmith RC, Dasovich M, Wang T, Guttler F, Woo SLC: A prevalent missense mutation in Northern Europe associated with hyperphenylalaninemia. Eur J Pediatr
1991;150:347–352 Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank the staff of the Child Development Institute at the Sheba Medical Center for their help
throughout this study, and the patients and their families for their cooperation. We are grateful to Dr. C.R. Scriver and his staff for compiling and distributing the PAH Mutation Consortium
lists. SLCW is an investigator at the Howard Hughes Medical Institute. This study was supported by the US-Israel Binational Science Foundation (BSF). AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Department of Human Genetics, Sackler School of Medicine, Tel Aviv University, Ramat Aviv, 69978, Israel Sandra Kleimam, Smadar Avigad, Lina Vanagaite, Miriam David &
Yosef Shiloh * Department of Middle Eastern and African History, Tel Aviv University, Ramat Aviv, Israel Aryeh Shmuelevitz * Department of Cell Biology and Institute of Molecular Genetics,
Baylor College of Medicine, Howard Hughes Medical Institute, Houston, Tex., USA Randy C. Eisensmith & Savio L. C. Woo * Child Development Institute, Sheba Medical Center, Tel Hashomer,
Israel Nathan Brand & Gerard Schwartz * Unité de Recherches sur les Handicaps Génétique de l’Enfant, INSERM U-12, Hôpital des Enfants-Malades, Paris, France Françoise Rey & Arnold
Munnich Authors * Sandra Kleimam View author publications You can also search for this author inPubMed Google Scholar * Smadar Avigad View author publications You can also search for this
author inPubMed Google Scholar * Lina Vanagaite View author publications You can also search for this author inPubMed Google Scholar * Aryeh Shmuelevitz View author publications You can also
search for this author inPubMed Google Scholar * Miriam David View author publications You can also search for this author inPubMed Google Scholar * Randy C. Eisensmith View author
publications You can also search for this author inPubMed Google Scholar * Nathan Brand View author publications You can also search for this author inPubMed Google Scholar * Gerard Schwartz
View author publications You can also search for this author inPubMed Google Scholar * Françoise Rey View author publications You can also search for this author inPubMed Google Scholar *
Arnold Munnich View author publications You can also search for this author inPubMed Google Scholar * Savio L. C. Woo View author publications You can also search for this author inPubMed
Google Scholar * Yosef Shiloh View author publications You can also search for this author inPubMed Google Scholar RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE
THIS ARTICLE Kleimam, S., Avigad, S., Vanagaite, L. _et al._ Origins of Hyperphenylalaninemia in Israel. _Eur J Hum Genet_ 2, 24–34 (1994). https://doi.org/10.1159/000472338 Download
citation * Received: 09 July 1993 * Revised: 11 November 1993 * Accepted: 22 November 1993 * Issue Date: January 1994 * DOI: https://doi.org/10.1159/000472338 SHARE THIS ARTICLE Anyone you
share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the
Springer Nature SharedIt content-sharing initiative KEY WORDS * Hyperphenylalaninemia * Phenylketonuria * Phenylalanine hydroxylase * Haplotypes * Mutations * Jews * Palestinian Arabs
Trending News
Do people still celebrate their name days in france?IT IS A FRENCH TRADITION TO SEND PEOPLE GOOD WISHES ON THEIR ‘NAME DAY’. WE LOOK AT WHERE THIS CAME FROM AND WHETHER THE...
Video 'Big Little Lies' star Laura Dern teases possible return to 'Jurassic Park' - ABC NewsABC NewsVideoLiveShowsShopStream onLive UpdatesLive UpdatesTrump 2nd term Ukraine drone attack Trump tariffs Boulder att...
Susannah townsend calls time on her international hockey career | england hockeyAfter playing 188 games across a 13-year career, Susannah Townsend has retired from international hockey. A double Olymp...
Royal heartbreak: how diana ‘struggle to cope' before deathThe Princess of Wales married Prince Charles in 1981, but sisters Lady Sarah McCorquodale and Baroness Jane Fellowes rea...
Page Not Found很抱歉,你所访问的页面已不存在了。 如有疑问,请电邮[email protected] 你仍然可选择浏览首页或以下栏目内容 : 新闻 生活 娱乐 财经 体育 视频 播客 新报业媒体有限公司版权所有(公司登记号:202120748H)...
Latests News
Origins of hyperphenylalaninemia in israelABSTRACT Mutations and polymorphisms at the phenylalanine hydroxylase (PAH) gene were used to study the genetic diversit...
Clinical risk factors and prognostic model for patients with bronchiolitis obliterans syndrome after hematopoietic stem cell transplantationABSTRACT Bronchiolitis obliterans syndrome (BOS) is a common and potentially devastating noninfectious pulmonary complic...
Cloud foundry | rcr wireless newsEditorial Report: Supporting AI, Using AI: Getting to pervasive intelligence in telecom networks Report...
Mitochondrial ros in cancer: initiators, amplifiers or an achilles' heel?KEY POINTS * Mitochondria contribute to the generation of ATP through oxidative phosphorylation, but they also participa...
Sridhar babu accuses brs of contradictory stance on ed; alleges secret ties with bjpHYDERABAD: IT and Industries Minister D Sridhar Babu accused the BRS of adopting contradictory approaches towards the En...